










 Servicios personalizados
Servicios personalizados
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroduction
During the past decades there has been a significant increase in our knowledge about the physiopathology of neurological conditions. Within these, diseases with immunological origin are responsible for major morbidity and even mortality, affecting the central nervous system (CNS) (multiple sclerosis (MS), neuromyelitis optica (NMO), limbic encephalitis…) and/or the peripheral system (Guillain-Barré syndrome (GBS), myasthenia gravis…) (Table 1). Overall, all these nosological conditions cause a major impact on patients and those around them but, given that some of them have a high prevalence, there can also be an impact for the public health system.

The constant study of new treatment targets has allowed a more efficient treatment of neurological diseases with immunological origin, with medications more specific for each condition, such as the case of MS, but it has also led to widening the range of diseases that can be potentially treated in this way. Thus, a deeper knowledge of the physiopathological mechanisms of conditions so prevalent as Alzheimer’s disease or migraine, so far not included in the group of “immune - mediated diseases”, has shown the role of immunomodulation treatments for their management, not only as hypotheses for the future but also as a short-term reality.
This article is intended as a brief review of the current situation of immunotherapy in neurological conditions, and to present some of the new therapeutic targets that will be offered to us in the near future.
Multiple sclerosis
MS is an inflammatory and demyelinating condition of the CNS. Its prevalence in Spain is of 91.2/100,0001, and there has been a confirmed increase in its prevalence during the past decades. It presents two evolution forms. The relapsing remitting form (RRMS) consists in the presence of relapses of focal and acute CNS inflammation which cause new symptoms, with or without irreversible accumulated sequelae (and without any clinical worsening when there are no relapses). The progressive form is defined by a worsening in the clinical status of the patient when there are no relapses. The treatment of the disease, besides symptomatic treatment and physiotherapy, is divided into the management of relapses and the use of “disease modifying drugs”.
Relapses are treated with glucocorticoids at high doses in short courses, typically 1 daily gram of intravenous methylprednisolone during 3 to 5 days. A multicenter, randomized, double-blind controlled clinical trial confirmed the non-inferiority of oral methylprednisolone 1,000 mg/day/3 days compared with intravenous methylprednisolone 1,000 mg/day/3 days2; therefore, oral treatment could be an alternative option to intravenous administration. In cases resistant to corticoid therapy, a plasmapheresis course will be effective in 72% of cases3.
Disease-modifying treatment is intended to improve the functional prognosis of patients at medium and long term. Until the launch of immuno-modulatory treatments specific for MS management, treatment consisted in immunosuppressants used for other conditions. In fact, azathioprine and mitoxantrone have the approved indication for their use in this disease. When interferon-beta 1b appeared in the 90’s decade, there was a revolution in MS treatment. Currently we have 17 different products for treatment of relapsing remitting MS, three for the treatment of secondary progressive forms, and one recently approved for the treatment of the primary progressive disease. All these have demonstrated their efficacy in randomized and controlled clinical trials, and this has led to the approval of the specific indication in their product specifications.Figure 1shows the molecules used for treatment of MS, according to their order of launch. It is worth highlighting the increasing frequency with which new molecules for this indication are being approved by regulatory agencies.

Figure 1. Molecules approved for Multiple Sclerosis treatment in chronological order of availability for clinical practice.
Both interferons-β and glatiramer acetate are being used for the past two decades with moderate efficacy and a verified safety profile, making them adequate as first line treatment. There is only correct acceptance by patients, because the way of administration is subcutaneous or intramuscular. For this reason, there has been a search for formulations with lower frequency of administration, such as pegylated interferon-β or glatiramer acetate 40 mg. Widely used, this type of medications is not always effective or well tolerated in all patients; therefore, research has focused on finding medications which are more potent and ways of administration which are more convenient for patients.
Teriflunomide and dimethyl fumarate, which are oral medications, currently allow treating MS patients without using parenteral administration. Teriflunomide4acts by a reversible inhibition of the dihydroorotate dehydrogenase enzyme, highly expressed in activated lymphocytes, causing a reduction in the proliferation of activated lymphocytes T and B. Terifluno-mide has demonstrated efficacy in RRMS treatment both in controlled and randomized clinical trials vs. placebo and in the clinical trial vs. interferon β1a 44 μg three times per week. Its safety profile is favorable but there is potential liver toxicity, which requires strict monitoring. It cannot be used in pregnant women until two years after discontinuation, because there is high teratogenic risk; there is a procedure for fast elimination which allows to reduce the product concentration below the risk considered minimal for the fetus (<0.02 μg/mL). Dimethyl fumarate5, with a mechanism of action still not completely understood, has demonstrated its efficacy in two clinical trials. Its gastrointestinal tolerability can be troublesome, unlike its other characteristic side effect: flushing. Some cases of progressive multifocal leukoencephalopathy (PML) have been published in association with this drug, which forces to conduct a periodical monitoring of the total lymphocyte count; it is recommended to interrupt the use of dimethyl fumarate if this count goes below 500/μL persistently during 6 continuous months. In terms of finding more effective drugs, medications such as alemtuzumab, fingolimod and natalizumab are those associated with higher efficacy for preventing relapses; natalizumab seems to be associated to a higher extent with a lower progression of disability6. Natalizumab is a recombinant humanized monoclonal antibody which binds with integrin alpha-4 beta-1 and blocks the interaction with the vascular cell adhesion molecule 1 (VCAM-1), thus preventing the migration to the CNS of mononuclear leukocytes through the endothelium of the blood-brain barrier. Its use has been associated with the development of PML in patients with positive test results for anti-JC virus antibodies, and this risk will be higher with a longer time of treatment, and also if the patient was treated with immunosuppressants before natalizumab. This fact is a major limitation for the use of this medication. Alemtuzumab has been evaluated in three clinical trials vs. an active comparator: interferon β1a 44 μg three times per week. A Cochrane evaluation reached the conclusion that alemtuzumab reduces the percentage of patients who suffer relapses, disability progression and development of new lesions, as seen in magnetic resonance imaging throughout 24 to 36 months, compared with interferon β7. Alemtuzumab has not been associated with PML development; but there have been potentially severe reactions to the intravenous infusion, infections, and autoimmune events which require strict monitoring. Fingolimod is a sphingosine-1-phosphate receptor modulator with oral administration, which has demonstrated its efficacy and safety for RRMS treatment in three Phase III clinical trials. It is more effective than weekly interferon β 30 μg in the reduction of relapse parameters and magnetic resonance imaging. Given that fingolimod also interacts with different subtypes of sphingosine-1-phosphate receptors (S1PR1, S1PR2, S1PR3, S1PR4, S1PR5), there is a risk of brachycardia and QT-interval prolongation, which requires patient monitoring during the first dose. An increase in blood pressure has been described, as well as macular edema, liver toxicity and some cases of PML, which require a careful monitoring of patients8. With the aim to improve the safety profile of sphingosine-1-phosphate receptor modulators, there are various molecules under research with higher selectivity for the S1PR1 receptor, which is the cause of the effect on lymphocytes, and has no effects on other organs or systems. The agents currently under development are: siponimod, ponesimod, ozanimod, ceralifimod, GSK2018682 and MT-1303. Siponimod stands out among these, because it has demonstrated, in a Phase III clinical trial on the secondary progressive form of MS vs. placebo, its ability to reduce the confirmed progression disability at 3 and 6 months by 21% and 26%, respectively9. These results could lead siponimod to become, alongside interferon β 1a 44 μg, interferon β1b and mitoxantrone, the fourth medication with specific indication for the treatment of secondary progressive MS.
In this overview of the near future of MS treatment, we cannot leave out two medications recently approved by the European Medicines Agency: ocrelizumab and oral cladribine. Ocrelizumab is a humanized anti-CD20 monoclonal antibody that has demonstrated high efficacy in RRMS treatment, based in two randomized and controlled clinical trials. It has also confirmed, and this is a milestone in MS treatment, its ability to delay the accumulated disability in patients with primary progressive MS10. Thus, it has the indication for RRMS but also for the treatment of those adult patients with early primary progressive MS who present inflammatory activity in imaging tests. Other anti-CD20 antibodies have already been used off-label for RRMS treatment (rituximab), or are currently in the stage of clinical development (ofatumumab). Oral cladribine11causes gradual lymphocyte depletion over the weeks, not associated with cell lysis, with higher impact on B cells than on T cells, and with reconstitution of the count of said cell lines throughout the months. In this way, when administered in two bimonthly cycles separated by one year, it acts as an inductor drug, not causing the prolonged immunosuppression of other previously mentioned medications which require uninterrupted treatment. The efficacy of cladribine has been demonstrated in two randomized and controlled Phase III clinical trials. Its main side effect is lymphopenia, associated with its mechanism of action; but it does not seem to be associated with an increase in neoplasia or infections vs. the control, except for the case of herpes zoster infections. Cladribine is indicated for adult patients with recurrent MS with high clinical or radiological activity.
The way of administration, potential effects, and recommended monitoring for the medications mentioned in this review appear summarized inTable 2.
Table 2. Immunomodulatory treatments: way of administration, potential risks, and recommended monitoring.
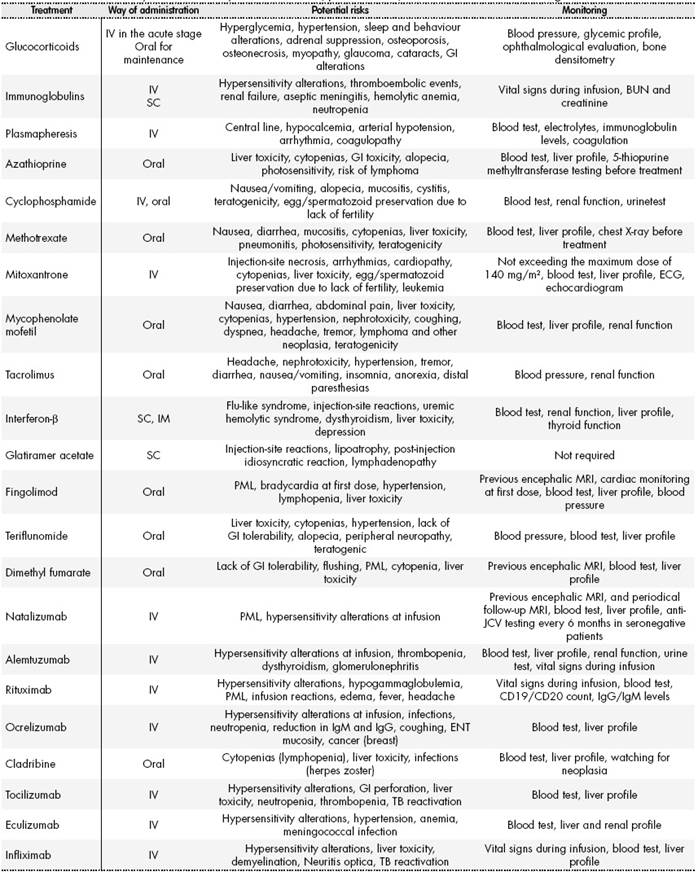
ABUN: Blood urea nitrogen test; MRI: Magnetic resonance imaging; GI: Gastrointestinal; TB: Tuberculosis; PML: Progressive multifocal leukoencephalopathy; ENT: Ear, nose and throat; ECG: Electrocardiogram; IgG: Immunoglobulin G; IgM: Immunoglobulin M; Anti-JCV: JC virus antibody test.
Neuromyelitis optica
NMO is an inflammatory demyelinating condition, anti-aquaporin 4 antibody - mediated (NMO-IgG). It affects specifically the spinal cord and the optic nerves. Its prevalence in Spain is of 1-5/100,000 inhabitants, and therefore less frequent than MS, but potentially more severe in the majority of cases.
Treatment of inflammatory relapses is conducted with 1 gram of intravenous methylprednisolone per day during 3 to 5 days, though its evidence comes from studies with MS or optic neuritis patients. Those patients who show no improvement with the previous regimen are treated with plasmapheresis12or intravenous human immunoglobulin13.
Regarding maintenance treatment, intended to prevent new relapses and accumulated disability, treatment will be typically initiated with azathioprine or mycophenolate mofetil, while the patient receives treatment with IV methylprednisolone, due to the time that these medications will take to start acting. Another first line option is rituximab14. Methotrexate would be reserved for those patients who don’t tolerate the previous treatments, or those for whom these are not effective15. Other potential treatments, but more dubious in terms of efficacy or toxicity, are tacrolimus, cyclosporine, mitoxantrone, and cyclophosphamide16. It is worth pointing out here that some disease-modifying drugs for MS, such as interferon β17, natalizumab18, and fingolimod19, will lead to a worsening in the evolution of NMO; therefore, it is essential to conduct an adequate differential diagnosis between both nosological entities.
In terms of new treatment options, various monoclonal antibodies are being evaluated for their use in NMO. Tocilizumab is a recombinant humanized anti-IL-6, which causes deletion of plasmablasts (which are CD20-, and therefore are not affected by rituximab); it has shown efficacy in some isolated cases20 21 22 23-24and in a Phase IV study25. On the other hand, eculizumab inhibits the complement pathway, preventing the cleavage of C5 into C5a and C5b, and therefore preventing the formation of membrane attack complex (C5b-C9). This molecule has shown potential for NMO treatment in a pilot open study with 14 patients26.
Other potentially valid treatment options, still pending adequate assessment, would be: aquaporumab (a humanized NMO-IgG monoclonal antibody with high affinity which might prevent pathogenic aquoporine-4 from binding to the Fc fragment), alemtuzumab (anti-CD52 previously described in the treatment for MS), and infliximab (chimeric monoclonal antibody anti-TNFα)27.
Migraine
Migraine is a highly prevalent neurological disease; 14.7% of the world population suffers it. It is the third human condition more frequent after tooth cavities and tension headache28. Its treatment is based on stopping the acute pain attack (with non-steroid anti-inflammatories and triptans, mainly); but also on medications to prevent new episodes, in those patients where the frequency and intensity of pain will justify their use. Currently, anti-hypertension drugs are used (betablockers), as well as antidepressants (tricyclics) and antiepileptic agents (topiramate, valproic acid), among others29. In some cases, botulinum toxin has also been used30 31-32.
In the past decade, blocking the calcitonin gene-related peptide (CGRP) has also been put forward as a new treatment target for preventing migraine episodes. This is due to the finding that CGRP levels increased during migraine episodes, and were reduced after using triptans33. Moreover, the intravenous administration of CGRP caused migraine episodes in migraine patients34. Two types of drugs have been designed in order to modify this CGRP design: on one hand, the CGRP-receptor antagonists, the “gepant” class, and on the other hand, monoclonal antibodies targeted to CGRP or its receptor. All these have demonstrated efficacy in controlled and randomized clinical trials, with efficacy at least equal than that of current preventive treatments.Figure 2summarizes the efficacy in clinical trials of monoclonal antibodies as prophylaxis for episodic and chronic migraine35 36 37 38 39-40.
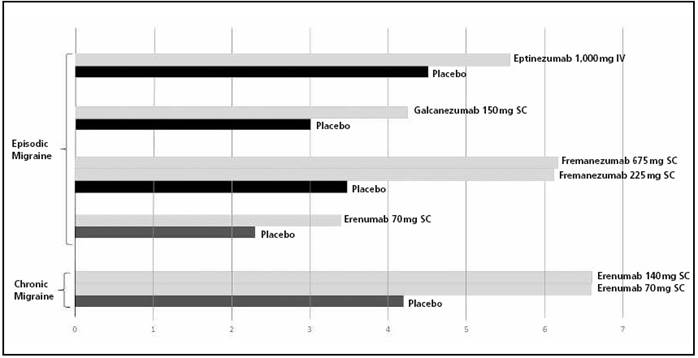
Figure 2. Efficacy of monoclonal antibodies in preventive treatment for migraine: reduction in migraine days per month (mean). All outcomes are statistically significant (p<0.05). IV: intravenous administration; SC: subcutaneous administration.
In terms of safety, liver toxicity problems have been observed with some gepants; this does not seem to happen with monoclonal antibodies41. Regarding the latter, in the absence of long-term data, some doubts can arise in terms of the cardiovascular system (hypertension, ischemic events), pituitary function, the GI system (constipation/diarrhea, ulcers, irritable bowel), and skin (erythema, inflammation, interference with wound healing)41. The potential presence of neutralizing antibodies which limit the effect of monoclonal antibodies over time, and the cost of treatment, can also be factors to take into account regarding their use in a disease with an extraordinary prevalence.
Movement disorders
Movement disorders caused by an autoimmune physiopathology can appear isolatedly or within a wider encephalopathic process including epileptic manifestations and/or cognitive deterioration. Traditionally, movement disorders have been classified into “hyperkinetic” (myoclonus, chorea, tics, pseudoathetosis, dystonia and other phenomena), and “hypokinetic” (Parkinsonism, stiff-person syndrome, progressive encephalomyelitis with rigidity and myoclonus). These are conditions with low prevalence (e.g. stiff-person: 1/1,250,000). In their treatment, besides determining if there is a causal neoplasia and eliminating it, treatment consists in the use of intravenous methylprednisolone, intravenous human immunoglobulin, or plasmapheresis in the acute stage, and azathioprine and mycophenolate mofetil as maintenance therapy42,43.
In this section, it is worth referring briefly to potential treatment targets in idiopathic Parkinson’s disease, a condition with a major prevalence (1-2/1,000)44. Physiopathological knowledge leads to consider that α-synuclein is a key molecule in neuronal death in Parkinson’s disease, by aggregating in toxic forms, spreading into the extracellular space and “contaminating” the adjacent neurons, thus perpetuating the pathogenic process. Thus, active or passive immunotherapy strategies intended to reduce the level of α-synuclein toxic extracellular aggregation could reduce or prevent the disease progression45,46.
Autoimmune epilepsy
Alongside epilepsy conditions associated with systemic autoimmune disorders, such as systemic lupus erythematosus, Hashimoto encephalopathy, sarcoidosis or celiac disease, there are antibody-mediated disorders which cause epileptic episodes as one of their main clinical manifestations. Said antibodies can be classified into those that bind intracellular antigens and those that bind to neuronal surface proteins. The first class includes the Hu, Ma2, CRMP5 and amphiphysin antibodies. Those conditions derived of intracellular antigens will present a worse response to immunotherapy47. Overall, immune treatment consists in a first line with intravenous methylprednisolone 500-1,000 mg/day/5 days, while intravenous immunoglobulin or plasmapheresis are reserved for steroid-resistant patients48. Second line treatment must be initiated within 2 weeks at most, if there has been no >50% reduction in episodes with first line agents48,49. The following will then be used: cyclophosphamide48, rituximab50, cyclophosphamide + rituximab, mycophenolate mofetil, or azathioprine48,49.
Dementia and autoimmune encephalopathies
The presentation form ranges from acute limbic encephalitis to subacute or chronic forms with a difficult differential diagnosis vs. primarily neuro-degenerative conditions. Their etiology is primarily idiopathic-autoimmune or in the context of a paraneoplastic phenomenon. In terms of treatment, the main objective in paraneoplastic conditions is to remove the causal tumour completely, whenever possible. For any of both physiopathological mechanisms, however, acute treatment of neurological symptoms will be, as in other autoimmune conditions, intravenous methylprednisolone used at high doses, or intravenous human immunoglobulin. Improvement with acute treatment can justify the use of maintenance therapy with corticosteroids, azathioprine, mycophenolate mofetil, methotrexate or tacrolimus, among other agents. In some conditions, such as anti-N-methyl-d-aspartate receptor encephalitis, drugs like rituximab or cyclophosphamide can be considered second-line treatment when there is little or no response with previously mentioned agents51,52. Acute disseminated encephalomyelitis, more frequent among pediatric patients, is a condition more typically monophasic, and therefore its management will usually be limited to acute treatment with intravenous methylprednisolone at high doses, intravenous human immunoglobulin, or plasmapheresis53.
Alzheimer’s disease
The prevalence of Alzheimer’s disease increases with age. In an estimation conducted for 2017 in the United States of America, 4% of people <65 -year-old present the disease, as well as 16% of persons between 65 and 74-year-old, 44% between 75 and 84-year-old, and 38% of people >85-year-old. It is expected that, at world level, the number of Alzheimer cases will triple by 205054. In an ageing society like ours in Spain, Alzheimer’s disease is already representing a major public health problem, and this will get even worse in the future.
The treatment for Alzheimer’s disease is based on those items considered key in its physiopathology, still only partly known (Table 3). A key event in this condition is the loss of cholinergic neurons, which has led to the prescription of cholinesterase inhibitors such as donepezil or rivastigmine. Another relevant element is the increase in glutamatergic activity, which has strengthened the indication of anti-N-methyl-D-aspartate receptor blockers such as memantine. Both therapeutic classes are typically used in current clinical practice. There are other three physiopathological mechanisms that can open new therapeutic targets, this time through the use of immunotherapies. The build-up of neuritic plaques (β-amyloid), neurofibrillary tangles (τ protein), and the local inflammation caused by microglia, are potential treatment objectives that are being evaluated in recent years55.
Table 3. Key physiopathological elements in Alzheimer’s disease and treatment strategies targeting them
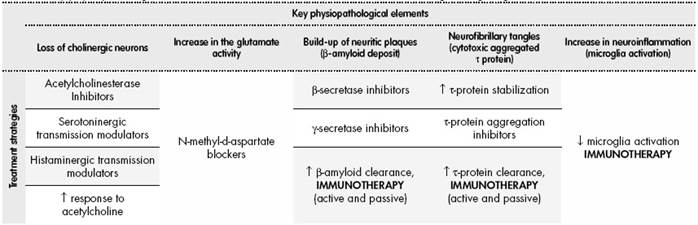
β-amyloid plaque is formed in two steps from the amyloid precursor protein (APP), through the β-secretase and γ- secretase enzyme complexes. It is thought that β-amyloid deposit could play a major role in the development of Alzheimer’s disease56. Keeping this hypothesis in mind, there has been a development, on one hand, of therapies able to reduce the activity of the β-secretase and γ-secretase complexes, with the objective of producing less β-amyloid; and, on the other hand, immunological treatments that can increase the elimination of the pathogenic β-amyloid already formed.
Thus, two strategies have been developed: active immunotherapy and passive immunotherapy. One of the first clinical trials on active immunotherapy was conducted with AN1792, a synthetic amyloid peptide (Aβ42) which induces the production of antibodies against the β-amyloid57. The Phase II clinical trial was stopped due to the development of meningoencephalitis in 6% of the study subjects. Besides, in those who did not develop meningoencephalitis, there was no demonstrated delay in the evolution of cognitive deterioration, despite a clear reduction in the senile plaque deposits. In view of these results, it was argued that the study included patients with moderate/severe Alzheimer’s disease; maybe treatment with drugs promoting the elimination of senile plaques could be indicated in earlier stages of the disease. With this recommendation, other molecules promoting active immunity to eliminate the amyloid plaques are being evaluated: among them, agent CAD106, safe and well tolerated, without any meningoencephalitis cases reported to date, is still under study58. Regarding passive immunotherapy, various molecules have been developed and are currently under evaluation. Bapineuzumab, targeting the N-terminal end of β-amyloid, has been discontinued due to its low efficacy and negative safety profile at Phase III59,60. On the other hand, solanezumab, which targets the monomer soluble β-amyloid, has presented an adequate safety profile, and it is being evaluated in patients with very early forms of Alzheimer’s disease. It has demonstrated an increase in β-amyloid-42 soluble protein in the cerebrospinal fluid of patients, with dose-dependent effect, which would support its mechanism of action of β-amyloid deposit elimination. However, it has not demonstrated clinical efficacy in mild Alzheimer’s disease61. Aducanumab is also on Phase III clinical trials, used for mild Alzheimer’s disease, but also for the mild cognitive impairment stage. Finally, gantenerumab, which interacts with β-amyloid fibrils to recruit microglia, activate phagocytosis and degrade neuritic plaques, is currently in early stages of clinical development; it has shown an adequate safety profile, and is pending efficacy outcomes in mild Alzheimer’s disease and mild cognitive impairment62.
Similarly as with neuritic plaques, the presence of neurofibrillary tangles formed by hyperphosphorilated τ protein seems to be one of the key elements responsible for pathogenesis in Alzheimer’s disease. Aggregated τ protein is cytotoxic; therefore, preventing its production or favoring its elimination could have a clinical effect on patients. As it has been mentioned for β-amyloid protein, there are molecules which could lead to the elimination of pathogenic τ protein, through active immunotherapy. AADvac-1, an active vaccine with a natural truncated form of τ, is currently on Phase II after having shown a good safety profile. C2N8E12 is a humanized anti-τ antibody currently on Phase II study, and safety and efficacy outcomes are expected for 202063.
Besides neuritic plaques (β-amyloid) and neurofibrillary tangles (τ), neuroinflammation is one of the key elements in the physiopathology of Alzheimer’s disease. Evident astrogliosis has been observed, among other signs of inflammation, around amyloid plaques, and various studies suggest a relationship between microglia activation, neuritic plaque formation, and the clinical progression of the disease. For this reason, a therapy targeted to inhibit the activation of microglia could be useful. However, negative results with tramiprosate (lack of efficacy at Phase III), ibuprofen and r-flurbiprofen, have reduced the expectations for this therapeutic group / mechanism of action. CHF 5074 is still on Phase II clinical trials: a microglia modulator for patients with mild cognitive impairment63.
Finally, it is worth pointing out that intravenous human immunoglobulin seems to be effective for maintaining the cognitive ability in patients with mild to moderate Alzheimer’s disease in Phase III clinical trials64.
Peripheral nervous system
-
- GBS: An acute sensory-motor polyradiculopathy, demyelinating, axonal or mixed, with inflammatory origin. The typical symptoms are parestesia, pain and loss of strength, though there are different clinical forms such as Miller Fisher syndrome, a sensory ataxia condition with involvement in the brain stem and oculomotor nerves. Its incidence is of 0.4 to 3.25 patients per 100,000 inhabitants and year65.
GBS is treated with plasmapheresis; its efficacy was confirmed in Cochrane’s 2012 review66. An alternative option is using intravenous human immunoglobulin at 0.4 g/kg/ day during 5 days. No placebo-controlled studies have been conducted, but a Cochrane review from 2014 confirmed its efficacy, comparable to plasmapheresis, after a systematic evaluation of 5 clinical trials67. Besides, two clinical trials confirmed similar efficacy but fewer side effects of immunoglobulin vs. plasmapheresis68,69. A complete review on this matter can be consulted in the article by Wijdicks and Klein70.
-
- Chronic Inflammatory Demyelinating Polyneuropathy (CIDP): Considered the chronic presentation (>8 weeks of duration) of the demyelinating form of GBS, its treatment also consists in the use of plasmapheresis or immunoglobulin. However, while glucocorticoids are considered ineffective in GMS, these are useful both in the acute stage and the maintenance treatment of CIDP71. Rituximab has shown efficacy in studies with small patient cohorts with this condition72,73. Eculizumab could be an option still not tested in CIDP. Fingolimod74and alemtuzumab75could play a role in the treatment of this disease, pending confirmation in randomized and controlled clinical trials.
- Multifocal motor neuropathy (MMN) is an autoimmune disorder with low prevalence (0.6-2 patients per 100,000 inhabitants)76. It causes a slowly progressive loss of strength, asymmetrical and mainly distal. MMN is mediated by antiganglioside antibodies, and it can be treated with human intravenous or subcutaneous immunoglobulin, thus improving its symptoms and preventing their progression. Four clinical trials77 78 79-80have demonstrated that 78% of patients treated with intravenous immunoglobulin improved significantly their motor ability vs. 4% treated with placebo. Even though the meta-analysis of said studies did not show significant differences in the improvement of disability81, the treatment guidelines by the European Federation of Neurological Societies (EFNS) recommend using 2 g/kg of intravenous human immunoglobulin for first line treatment of MMN; this dose must be administered over 2 to 5 days. These guidelines also state that the maintenance dose of intravenous human immunoglobulin to be administered after an initial improvement with the first cycle should be 1 g/kg every 2 to 4 weeks, or 2 g/kg every 1-2 months82. It is worth noting that subcutaneous immunoglobulin has demonstrated similar efficacy to the intravenous formulation in MMN treatment, both for early stages83and as maintenance84. For an extensive review about this, it is recommended to read the work by Kumar et al.85.
- Amyotrophic lateral sclerosis (ALS) is a degenerative motor neuron di-sease. Physiopathological mechanisms of immunological substrate have recently been mentioned, thus opening a potential pathway for immunotherapy treatment. A review on this topic86describes the use of treatments for Rheumatoid Arthritis: anakinra (a recombinant analog of the interleukin-1 receptor antagonist), with negative outcomes; mastinib (a tyrosine-kinase inhibitor), with an on - going Phase III clinical trial, and tocilizumab, also under clinical trial, in this case Phase II. Treatments for MS have also been used, such as glatiramer acetate (negative outcomes) or fingolimod (efficacy not demonstrated). The lack of efficacy outcomes obtained with intravenous immunoglobulin, celecoxib, ozanezumab, NP001 (taurine), thalidomide, granulocyte stimulating growth factor, cyclosporine, or total lymphoid irradiation, have not prevented continuing with the line of research of immunological therapies for ALS. Other agents, such as ibudilast (a TLR4 and phosphodiesterase 3 and 4 inhibitor), RNS60, or drugs used to prevent rejection in transplants (basaliximab + mycophenolate mofetil + tacrolimus + glucocorticoids) are currently under investigation.
- Myasthenia gravis I an antibody-mediated condition (anti-acetylcholine receptor -AChR- or anti-muscle specific kinase (MuSK)), which prevents an adequate transmission in the motor plaque. Its characteristic symptom is muscle fatigue. Initial treatment, besides acetylcholinesterase inhibitors, is based on the use of oral prednisone, intravenous immunoglobulin and/or plasmapheresis for disease relapses. In order to avoid the continuous use of glucocorticoids in patients with generalized disease, the following are used: azathioprine, mycophenolate mofetil, cyclosporine A, methotrexate, tacrolimus, or cyclophosphamide. For a complete review on the use of these medications for myasthenia gravis, the study by Lee and Jander87is recommended. Another treatment option is rituximab, an anti-CD20 chimeric monoclonal antibody, suggested for patients with moderate-severe forms of the disease who are refractory to other treatments, and for those who are anti-MuSK-positive88,89. A meta-analysis evaluating 15 non-controlled clinical trials, with 168 patients included in total, on different treatment regimens with rituximab, seems to show its efficacy in the treatment of AchR-positive, MuSK-positive and AchR/MuSK-double negative myasthenia gravis90. More recently, eculizumab has demonstrated efficacy in a Phase II clinical trial91and in another in Phase III92; for this reason, it could be considered initially as a treatment option for severe cases or those refractory to other treatment strategies93.
- Autoimmune myopathies: The prevalence of polymyositis and dermatomyositis, the two most frequent autoimmune myopathies, is of 21.5/100,000 inhabitants94. Corticosteroids are the first line treatment for both conditions, as well as for immune-mediated necrotizing myopathy95 96-97. In patients with severe symptoms (dysphagia or inability to walk), intravenous methylprednisolone is used at 1 g/day/3 days, followed by oral prednisone in a decreasing dose starting with 60 mg/ day of prednisone. In moderate cases, it is possible to initiate oral treatment without the previous intravenous loading dose. In milder clinical presentations, it is possible to start at lower prednisone doses. Once muscular strength returns to normal, a progressive dose reduction will be implemented. In patients with severe disease, those with incomplete response to corticosteroids after 2 months of treatment, and those where the dose cannot be reduced below 10 mg/day, it is recommended to use azathioprine, methotrexate or mycophenolate mofetil. If there is failure or lack of tolerability to these second line medications, it could be possible to resort to rituximab, cyclosporine, cyclophosphamide, TNFα blockers98. Intravenous human immunoglobulin is effective for dermatomyositis treatment99and possibly also for polymyositis. It can be used as second line for patients with severe symptoms, because its effect is faster than that of said treatment line. The typical dose is 2 g/kg, distributed between 3 to 5 days. Inclusion-body myositis does not respond to im-munotherapy.
Discussion
Currently, immunological treatments allow us to treat a high number of neurological conditions in a more accurate and individualized way. The launch of products designed with specific treatment targets and evaluated in clinical trials with high level of evidence has become more frequent in clinical practice. Moreover, the range of conditions that can be potentially treated with immunotherapy is increasingly higher, and has started to inclu-de highly prevalent neurological conditions such as migraine and, possibly, Alzheimer’s disease. The high impact that these therapies could entail for the public health system will require compromises and consensus by all actors involved in their adequate use.

