










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkFarmacia Hospitalaria
versión On-line ISSN 2171-8695versión impresa ISSN 1130-6343
Farm Hosp. vol.42 no.6 Toledo nov./dic. 2018 Epub 09-Nov-2020
https://dx.doi.org/10.7399/fh.11031
REVISIÓN
Inmunoterapia en enfermedades neurológicas, presente y futuro
1Servicio de Neurología, Hospital Universitario Fundación Jiménez Díaz UTE, Madrid. España.
Objetivo:
El objetivo del presente trabajo es resumir el tratamiento inmunológico de las enfermedades neurológicas, describiendo la situación actual y los retos y oportunidades que se presentan en un futuro próximo.
Método:
Se realiza un análisis bibliográfico para, tras clasificar topográficamente las patologías neurológicas autoinmunes, presentar las más relevantes según las opciones inmunoterapéuticas disponibles. Asimismo, se exponen otras enfermedades neurológicas que serán nuevas candidatas a terapia inmunológica en el futuro.
Resultados:
Existen múltiples patologías neurológicas con base autoinmune, aunque su fisiopatología, a veces, solo sea parcialmente conocida. Sin embargo, pocos son los ensayos clínicos aleatorizados y controlados que soportan la evidencia de los tratamientos inmunológicos con los que las tratamos. Esta situación está cambiando rápidamente en enfermedades como la esclerosis múltiple, donde ensayos clínicos con un nivel de evidencia grado 1 son la norma. La enfermedad de Alzheimer y la migraña son algunas de las más prevalentes que se están incorporando al grupo de candidatas a inmunoterapia.
Conclusiones:
Con un número rápidamente creciente de terapias inmunológicas y de enfermedades neurológicas potencialmente tratables por esta vía, será necesaria una adecuada evaluación del impacto sociosanitario que van a conllevar para llegar a compromisos y consensos por parte de todos los actores implicados en su manejo.
PALABRAS CLAVE: Inmunoterapia; Patologías neurológicas; Enfermedades autoinmunes; Esclerosis múltiple; Neuromielitis óptica; Enfermedad de Alzheimer; Migraña; Trastornos del movimiento
Introducción
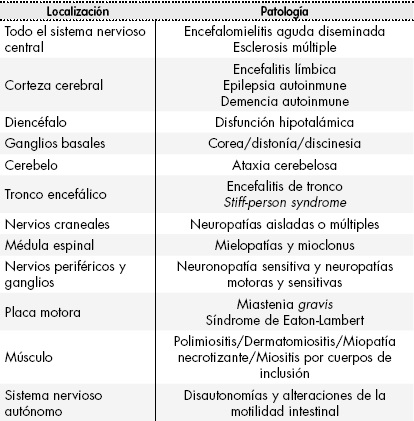
En las últimas décadas hemos asistido a un significativo crecimiento del conocimiento sobre la fisiopatología de las enfermedades neurológi-cas. Dentro de ellas, las patologías de origen inmunológico son respon-sables de una gran morbilidad e incluso mortalidad, por afectación del sistema nervioso central (SNC) (esclerosis múltiple (EM), neuromielitis óptica NMO), encefalitis límbica, etc.) y/o periférico (síndrome de GuillainBarré (SGB), miastenia gravis, etc.) (Tabla 1). En general, todas estas entidades nosológicas tienen un gran impacto para el paciente y sus allegados pero, dado que algunas son muy prevalentes, también pueden tenerlo para el sistema sociosanitario.
Tabla 1. Localización preferente de la patología neurológica de origen inmunológico y enfermedades asociadas
El constante estudio de nuevas dianas terapéuticas ha permitido tratar con mayor eficacia las enfermedades neurológicas de origen inmunoló-gico, con medicamentos más específicos para cada patología, como en el caso de la EM, pero también ampliar el espectro de enfermedades potencialmente abordables por esta vía. De esta forma, el conocimiento en mayor profundidad de los mecanismos fisiopatológicos de enfermedades tan prevalentes como la demencia de Alzheimer o la migraña, no incluidas hasta ahora en el grupo de “enfermedades inmunomediadas”, ha puesto de manifiesto el papel de los tratamientos inmunomoduladores en su manejo, no sólo como hipótesis de futuro sino como realidad a corto plazo.
En este trabajo pretendemos hacer una breve revisión del estado actual de la inmunoterapia en las enfermedades neurológicas y presentar algunas de las nuevas dianas terapéuticas que se nos ofrecerán en los próximos años.
Esclerosis múltiple
La EM es una enfermedad inflamatoria y desmielinizante del SNC. Su prevalencia en España es de 91,2/100.0001, constatándose un aumento en las últimas décadas. Presenta dos formas evolutivas. La EM recurrente remitente (EMRR) consiste en la aparición de “brotes” de inflamación focal y aguda del SNC que provocan nuevos síntomas con o sin acumulación irreversible de secuelas (y sin empeoramiento clínico en ausencia de brotes). La forma progresiva se define por el agravamiento del estado clínico del paciente en ausencia de brotes. El tratamiento de la enfermedad, además del abordaje sintomático y la fisioterapia, se divide en el manejo de los brotes y el uso de “modificadores del curso de la enfermedad”.
Los brotes se tratan con glucocorticoides a altas dosis en pulsos cortos, generalmente 1 gramo de metilprednisolona intravenosa al día durante 3 a 5 días. Un ensayo multicéntrico, aleatorizado, doble ciego y controlado confirmó la no inferioridad de la metilprednisolona oral 1.000 mg/día/3 días comparada con la metilprednisolona intravenosa 1.000 mg/día/3 días2, por lo que el tratamiento oral podría ser una alternativa a la vía intravenosa. En los casos resistentes a corticoterapia, un curso de plasmaféresis resulta eficaz en el 72% de las ocasiones3.
El tratamiento modificador del curso de la enfermedad pretende mejorar el pronóstico funcional de los pacientes a medio y largo plazo. Hasta la llegada de los tratamientos inmunomoduladores específicos para el manejo de la EM, el tratamiento consistía en el uso de inmunosupresores utilizados en otras patologías. De hecho, azatioprina y mitoxantrona tienen su indicación en ficha técnica para su uso en esta enfermedad. A partir de la aparición del interferón β1b en los años 90 del siglo xx, el tratamiento de la EM ha sufrido una revolución. Actualmente disponemos de 17 productos diferentes para el tratamiento de la enfermedad recurrente remitente, tres para el tratamiento de las formas secundariamente progresivas y uno recientemente aprobado para el tratamiento de la forma primariamente progresiva. Todos ellos han demostrado su eficacia en ensayos clínicos aleatorizados y controlados y, gracias a ello, cuentan con indicación específica en ficha técnica. En laFigura 1se presentan los principios activos utilizados para el tratamiento de la EM según el orden de aparición en el mercado. Nótese la frecuencia cada vez mayor con la que las nuevas moléculas para esta indicación van siendo aprobadas por las agencias reguladoras.

Figura 1. Moléculas aprobadas para el tratamiento de la esclerosis múltiple en orden cronológico de disponibilidad para la práctica clínica.
Tanto los interferones β como el acetato de glatiramero se utilizan desde hace dos décadas con una eficacia moderada y un perfil de seguridad contrastado, lo que los hace idóneos como primera línea de tratamiento. Su aceptación por parte de los enfermos es sólo correcta, ya que son administrados por vía subcutánea o intramuscular. Por este motivo, se han ido buscando formulaciones de administración menos frecuente, como el interferón β pegilado o como el acetato de glatiramero 40 mg. Ampliamente utilizados, esta clase de medicamentos no siempre es eficaz o bien tolerada en todos los pacientes, por lo que la investigación se ha volcado en encontrar medicamentos más potentes y vías de administración más cómodas para el paciente.
Los medicamentos orales teriflunomida y dimetilfumarato permiten, hoy día, tratar a los pacientes con EM sin tener que recurrir a la vía parenteral. La teriflunomida4actúa por vía de la inhibición reversible de la enzima dihidroorotato deshidrogenasa, altamente expresada en linfocitos activados, produciendo una reducción de la proliferación de linfocitos T y B activados. Tanto en los estudios controlados y aleatorizados contra placebo como en el realizado contra interferón β1a 44 μg tres veces por semana, teriflunomida ha demostrado su eficacia en el tratamiento de la EMRR. Su perfil de seguridad es favorable pero, debido a una potencial hepatotoxicidad, se requiere una monitorización analítica estricta. No se permite su uso en mujeres embarazadas hasta dos años después de cesar su uso por un riesgo teratogénico alto, existiendo un procedimiento de eliminación acelerada que permite reducir la concentración del producto por debajo del riesgo considerado mínimo para el feto (< 0,02 μg/ml). El dimetilfumarato5, con un mecanismo de acción todavía no enteramente conocido, ha demostrado su eficacia en dos ensayos clínicos. Su tolerancia gastrointestinal puede ser problemática, no tanto su otro efecto secundario característico, el flushing. Se ha publicado algún caso de leucoencefalopatía multifocal progresiva (LMP) con este fármaco, lo que obliga a una monitorización periódica del recuento de linfocitos totales, recomendándose la suspensión de dimetilfumarato si el recuento está por debajo de 500/μl de forma persistente durante 6 meses seguidos.
En el sentido de encontrar fármacos más eficaces, medicamentos como alemtuzumab, fingolimod y natalizumab son los que, en una reciente revisión, se asocian con una mayor eficacia para la prevención de brotes, siendo este último el que parece relacionarse en mayor medida con una menor progresión de la discapacidad6. Natalizumab es un anticuerpo recombinante monoclonal humanizado que se une a la integrina α4-β1 y bloquea la interacción con la molécula de adhesión de células vasculares 1 (VCAM -1), evitando así la migración de los leucocitos mononucleares a través del endotelio de la barrera hematoencefálica hacia el SNC. Su uso se asocia a la aparición de LMP en pacientes positivos para anticuerpos antivirus JC, siendo mayor este riesgo cuanto mayor sea el tiempo de tratamiento y también si el paciente fue, con anterioridad a natalizumab, tratado con inmunosupresores. Este hecho limita de forma importante el uso de este medicamento. Alemtuzumab ha sido evaluado en tres ensayos clínicos con un comparador activo, el interferón β1a 44 μg tres veces por semana. Una evaluación de la Cochrane concluye que alemtuzumab reduce la proporción de pacientes que sufren brotes, progresión de la discapacidad y desarrollo de nuevas lesiones en la resonancia magnética a lo largo de 24 a 36 meses, comparado con interferón β7. Alemtuzumab no se ha visto relacionado con el desarrollo de LMP, pero se asocia a reacciones a la infusión intravenosa, a infecciones y a eventos autoinmunes potencialmente graves que obligan a una monitorización estricta. Fingolimod es un modulador del receptor de la esfingosina-1-fosfato que, administrado por vía oral, ha demostrado mediante tres ensayos clínicos de fase 3 su eficacia y seguridad en el tratamiento de la EMRR. Es más eficaz que interferón β 30 μg a la semana en la reducción de los parámetros de brotes y resonancia magnética. Dado que fingolimod interactúa también con receptores de la esfingosina-1-fostato de diversos subtipos (S1PR1, S1PR2, S1PR3, S1PR4, S1PR5), existe un riesgo de bradicardia y prolongación del intervalo QT que obliga a monitorizar al paciente durante la primera dosis. Se ha descrito incremento de la tensión arterial, edema macular, toxicidad hepática y algunos casos de LMP, lo que obliga a una monitorización cuidadosa de los pacientes8. Intentando mejorar el perfil de seguridad de los moduladores del receptor de la esfingosina-1-fosfato, están en fase de investigación varias moléculas con mayor selectividad por el receptor S1PR1, responsable del efecto sobre los linfocitos y exento de efectos sobre otros órganos y sistemas. Los agentes actualmente en desarrollo son siponimod, ponesimod, ozanimod, ceralifimod, GSK2018682 y MT-1303. Cabe destacar, entre todos ellos, el siponimod, por haber demostrado en un ensayo clínico de fase 3 en la forma secundariamente progresiva de la EM que es capaz de reducir, con respecto a placebo, la progresión de la discapacidad confirmada a tres meses y seis meses en un 21% y un 26%, respectivamente9. Estos resultados podrían hacer que siponimod pasase a ser, junto con el interferón β1a 44 μg, el interferón β1b y la mitoxantrona, el cuarto medicamento con indicación específica parar el tratamiento de la EM secundariamente progresiva.
En esta mirada hacia el futuro próximo del tratamiento de la EM, no podemos dejar de referirnos a dos medicamentos recientemente aprobados por la Agencia Europea del Medicamento, ocrelizumab y cladribina oral. Ocrelizumab es un anticuerpo monoclonal anti-CD20 humanizado que ha demostrado una alta eficacia en el tratamiento de la EMRR, basado en dos ensayos clínicos aleatorizados y controlados. También ha confirmado, y esto es un hito en el tratamiento de la EM, que es capaz de retrasar la acumulación de la discapacidad en pacientes con EM primariamente progresiva10. Así, tiene indicación para la EMRR pero también para el tratamiento de pacientes adultos con EM primaria progresiva temprana y que presenten actividad inflamatoria en las pruebas de imagen. Otros anticuerpos anti-CD20 ya han sido utilizados para el tratamiento de la EMRR, fuera de indicación de ficha técnica (rituximab) o se encuentran en fase de desarrollo clínico (ofatumumab). Cladribina oral11produce una depleción de linfocitos gradual, a lo largo de semanas, no asociada a lisis celular,
con mayor impacto en células B que T, y con una reconstitución de los recuentos de dichas estirpes celulares a lo largo de meses. De esta forma, administrado en dos ciclos bimensuales separados un año, actúa como un fármaco inductor, sin producir la inmunosupresión prolongada de otros medicamentos nombrados anteriormente y que requieren un tratamiento ininterrumpido. La eficacia de cladribina ha sido demostrada en dos ensayos clínicos aleatorizados y controlados de fase 3. Su efecto secundario principal es la linfopenia, relacionada con su mecanismo de acción, pero no parece asociarse a un aumento de neoplasias ni de infecciones con respecto al control, salvo en el caso de las infecciones por herpes zóster. Cladribina está indicada en pacientes adultos con EM recurrente de alta actividad clínica o radiológica.
La vía de administración, los efectos potenciales y la monitorización recomendada de los medicamentos nombrados en esta revisión se encuentran resumidos en laTabla 2.
Tabla 2. Tratamientos inmunomoduladores: vía de administración, riesgos potenciales y monitorización recomendada.
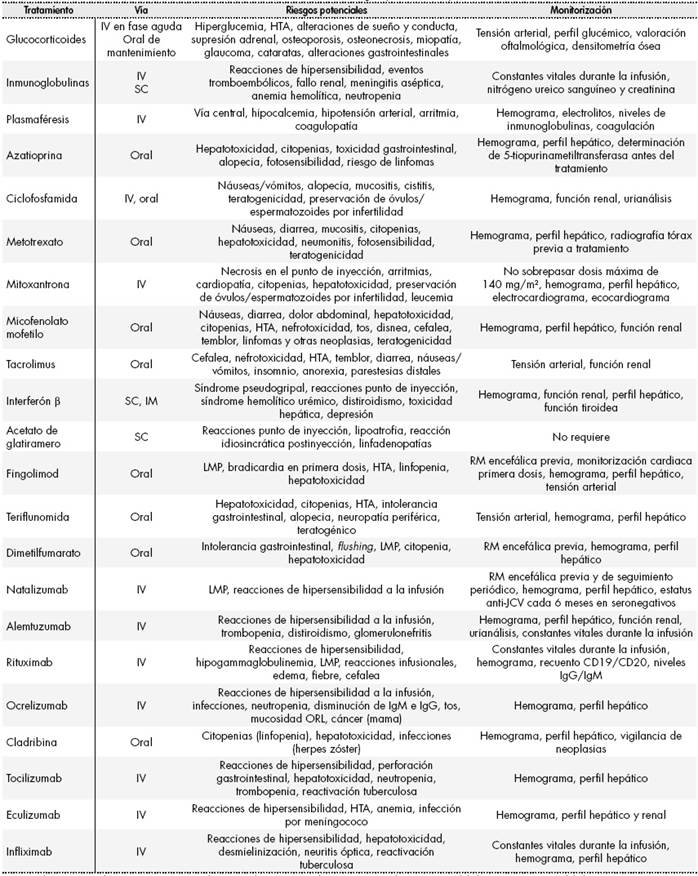
Anti-JCV: determinación de anticuerpos anti virus John Cunningham (JC); HTA: hipertensión arterial; IgG: inmunoglobulina G; IgM: inmunoglobulina M; LMP: leucoencefalopatía multifocal progresiva; ORL: otorrinolaringológico; RM: resonancia magnética.
Neuromielitis óptica
La NMO es una patología inflamatoria desmielinizante mediada por el anticuerpo antiaquaporina 4 (NMO-IgG). Afecta específicamente a la médula espinal y los nervios ópticos. Con una prevalencia en España de 1-5/100.000 habitantes, es menos frecuente que la EM pero potencialmente más grave en la mayoría de los casos.
El tratamiento de los brotes inflamatorios se realiza con 1 gramo de metilprednisolona intravenosa al día durante tres a cinco días, aunque la evidencia proviene de estudios de pacientes con EM o neuritis ópticas. En aquellos pacientes que no mejoran con la pauta anterior, se recurre a la plasmaféresis12o al uso de inmunoglobulina humana intravenosa13.
En cuanto al tratamiento de mantenimiento, con la intención de evitar nuevos brotes y la acumulación de discapacidad, se suele iniciar tratamiento con azatioprina o micofenolato mofetilo mientras el paciente recibe el tratamiento con metilprednisolona intravenosa, debido al tiempo que tardan en comenzar a hacer efecto estos medicamentos. Otra opción de primera línea es el rituximab14. El metotrexato quedaría reservado para los pacientes que no toleran o en los cuales no son eficaces los anteriores tratamientos15. Otros tratamientos potenciales, pero sobre los que existen más dudas en cuanto a la eficacia o la toxicidad, son tacrolimus, ciclosporina, mitoxantrona y ciclofosfamida16. Cabe aquí señalar que algunos medicamentos modificadores de la evolución de la EM, como interferón β17, natalizumab18y fingolimod19, empeoran el curso de la NMO, por lo que un adecuado diagnóstico diferencial entre las dos entidades nosológicas es fundamental.
En cuanto a nuevas opciones terapéuticas, se están evaluando varios anticuerpos monoclonales para su uso en la NMO. El tocilizumab, un anticuerpo recombinante humanizado bloqueador de la interleucina-6 que produce una depleción de plasmablastos (que son CD20- y por lo tanto no se ven influidos por el rituximab) ha mostrado eficacia en casos aislados20 21 22 23-24y en un estudio de fase 425. Por su parte, el eculizumab inhibe la vía del complemento impidiendo la escisión de C5 a C5a y C5b y, por ese motivo, impide la formación del complejo de ataque a la membrana (C5b-C9). Esta molécula ha mostrado potencial en el tratamiento de la NMO en un estudio piloto y abierto de 14 pacientes26.
Otras opciones terapéuticas potencialmente válidas pero pendientes de una adecuada evaluación serían el aquaporumab (anticuerpo monoclonal humanizado NMO-IgG de alta afinidad que podría impedir la unión de la aquoporina-4 patógena al fragmento Fc), el alemtuzumab (anti-CD52 descrito previamente en el tratamiento de la EM) y el infliximab (anticuerpo monoclonal quimérico anti-TNFα)27.
Migraña
La migraña es una enfermedad neurológica de altísima prevalencia. Un 14,7% de la población mundial la sufre. Es la tercera patología humana más frecuente tras la caries y la cefalea tensional28. Su tratamiento se basa en el cese del brote agudo de dolor (con antiinflamatorios no esteroideos y triptanes, principalmente) pero también, en aquellos pacientes en que la frecuencia y la intensidad de los dolores lo justifica, en el uso de medicamentos preventivos de nuevas crisis. A día de hoy, estos últimos provienen de fármacos contra la hipertensión (betabloqueantes), la depresión (antidepresivos tricíclicos) y la epilepsia (topiramato, ácido valproico), entre otros29. En algunos casos también se utiliza la toxina botulínica30 31-32.
En la última década, el bloqueo del péptido relacionado con el gen de la calcitonina (CGRP en su acrónimo en inglés) ha sido postulado como una nueva diana terapéutica para la prevención de las crisis de migraña. Esto se debe al hallazgo de que los niveles de CGRP se incrementaban durante las crisis de migraña y se veían reducidos tras el uso de triptanes33. Además, la administración intravenosa de CGRP provocaba ataques de migraña en pacientes migrañosos34. Para modificar esta respuesta CGRP se han diseñado dos clases de fármacos. Por un lado, los antagonistas del receptor CGRP, la familia de los “gepantes”. Por otro, los anticuerpos monoclonales dirigidos al CGRP o a su receptor. Todos ellos han demostrado eficacia en ensayos clínicos controlados y aleatorizados, con una eficacia al menos igual que las de los tratamientos preventivos actuales. En laFigura 2se resume la eficacia en ensayos clínicos de los anticuerpos monoclonales en la prevención de la migraña episódica y crónica35 36 37 38 39-40.
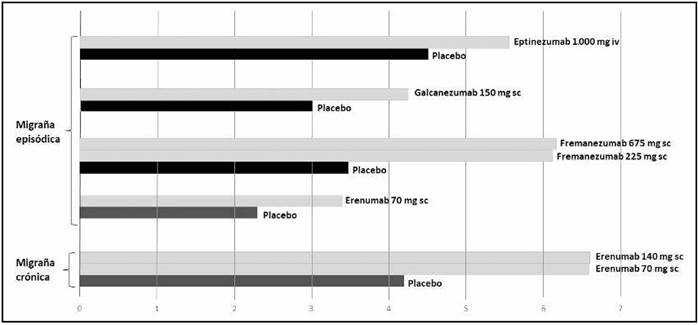
Figura 2. Eficacia de los anticuerpos monoclonales en el tratamiento preventivo de la migraña: reducción de días de migraña por mes (media). Todos los resultados son estadísticamente significativos (p < 0,05). IV: administración intravenosa; SC: administración subcutánea.
En cuanto a la seguridad, se han visto problemas de toxicidad hepática con alguno de los gepantes, lo que no parece suceder con los anticuerpos monoclonales41. En relación con estos últimos, en ausencia de datos a largo plazo, algunas dudas se pueden plantear en cuanto al sistema cardiovascular (hipertensión arterial (HTA), eventos isquémicos), a la función pituitaria, al sistema gastrointestinal (estreñimiento/diarrea, ulcus, colon irritable) y a la piel (eritema, inflamación, interferencia en la cicatrización de heridas)41. La posible presencia de anticuerpos neutralizantes que limiten en el tiempo el efecto de los anticuerpos monoclonales y el coste del tratamiento pueden ser factores a tener también en consideración respecto a su uso en una enfermedad extraordinariamente prevalente.
Trastornos del movimiento
Los trastornos del movimiento provocados por una fisiopatología autoinmune pueden aparecer de forma aislada o dentro de un proceso encefalopático más amplio que incluya manifestaciones epilépticas y/o deterioro cognitivo. Clásicamente, los trastornos del movimiento se clasifican en “hipercinéticos” (mioclonus, corea, tics, pseudoatetosis, distonía y otros fenómenos) e “hipocinéticos” (parkinsonismo, stiff-person syndrome, encefalomielitis progresiva con rigidez y mioclonus). Son patologías de muy baja prevalencia (stiff-person 1/1.250.000). En su tratamiento, aparte de determinar si existe una neoplasia causal y de erradicarla, el tratamiento pasa por el uso de metilprednisolona intravenosa, inmunoglobulina humana intravenosa o plasmaféresis en la fase aguda, y por azatioprina y micofenolato mofetilo como terapia de mantenimiento42,43.
Cabe en este apartado hacer una breve referencia a posibles dianas terapéuticas en la enfermedad de Parkinson idiopática, una enfermedad de importante prevalencia (1-2/1.000)44. Los conocimientos fisiopatológicos llevan a considerar la α-sincleína una molécula clave en la muerte neuronal en la enfermedad de Parkinson al agregarse en formas tóxicas, salir al espacio extracelular y “contaminar” las neuronas adyacentes, perpetuando el proceso patogénico. De esta forma, estrategias de inmunoterapia activa o pasiva que pretendan disminuir el nivel de agregados extracelulares tóxicos de α-sincleína podrían reducir o evitar la progresión de la enfermedad. Los diversos productos en estudio se encuentran aún en fases 1 y 2 de desarrollo clínico45,46.
Epilepsia autoinmune
Además de las epilepsias relacionadas con trastornos autoinmunes sistémicos tales como el lupus eritematoso sistémico, la encefalopatía de Hashimoto, la sarcoidosis o la celiaquía, hay trastornos mediados por autoanticuerpos que producen cuadros epilépticos como una de sus principales manifestaciones clínicas. Dichos anticuerpos se pueden clasificar en aquéllos que fijan antígenos intracelulares y aquéllos que se unen a proteínas de superficie neuronal. Entre los primeros se encuentran los anticuerpos Hu, Ma2, CRMP5 y anfifisina. Son las patologías derivadas de los antígenos intracelulares las que peor van a responder a la inmunoterapia47. En general, el tratamiento inmune se basa en una primera línea formada por metilprednisolona intravenosa 500-1.000 mg/día/5 días, quedando la inmunoglobulina intravenosa o la plasmaféresis para pacientes resistentes a los esteroides48. El tratamiento de segunda línea se debe iniciar en el plazo máximo de dos semanas si no ha habido una reducción superior al 50% de las crisis con los agentes de primera línea48,49. Se utilizan entonces ciclofosfamida48, rituximab50, ciclofosfamida + rituximab, micofenolato mofetilo o azatioprina48,49.
Demencia y encefalopatías autoinmunes
Sus formas de presentación varían desde la encefalitis límbica aguda a formas subagudas o crónicas de difícil diagnóstico diferencial con los procesos primariamente neurodegenerativos. Su etiología es primariamente idiopática autoinmune o en el contexto de un fenómeno paraneoplásico. En cuanto al tratamiento, en el caso de los procesos paraneoplásicos, el objetivo principal es, si resulta posible, retirar por completo el tumor causal. Para cualquiera de los dos mecanismos fisiopatológicos, sin embargo, el tratamiento agudo de los síntomas neurológicos es, como en otras patologías autoinmunes, el uso de metilprednisolona intravenosa en altas dosis o de inmunoglobulina humana intravenosa. La mejoría con el tratamiento agudo puede justificar el uso de una terapia de mantenimiento con corticoides, azatioprina, micofenolato mofetilo, metotrexato o tacrolimus, entre otros agentes. En algunas patologías, como la encefalitis antirreceptor de N-metil-D-aspartato, agentes como el rituximab o la ciclofosfamida pueden considerarse de segunda línea cuando no hay respuesta o ésta es escasa a los agentes mencionados con anterioridad51,52. La encefalomielitis aguda diseminada, más frecuente en la edad pediátrica, es una enfermedad más comúnmente monofásica, por lo que el tratamiento suele limitarse al tratamiento agudo con metilprednisolona intravenosa en altas dosis, inmunoglobulina humana intravenosa o plasmaféresis53.
Enfermedad de Alzheimer
La prevalencia de la enfermedad de Alzheimer se incrementa con la edad. En una estimación realizada para los Estados Unidos de América para el año 2017, presentan la enfermedad el 4% de los menores de 65 años, el 16% de las personas entre 65 y 74 años, el 44% entre 75 y 84 años, y el 38% en los mayores de 85 años. Se espera que, a nivel mundial, el número de casos de Alzheimer se triplique para el año 205054. En una sociedad que envejece como la española, la enfermedad de Alzheimer está suponiendo ya, y supondrá aún más en el futuro, un importante problema sociosanitario.
El tratamiento de la enfermedad de Alzheimer se basa en los puntos considerados clave de su fisiopatología, todavía sólo en parte conocida (Tabla 3). Un evento clave en esta patología es la pérdida de neuronas colinérgicas, la cual ha promovido la prescripción de anticolinesterásicos tales como el donepezilo o la rivastigmina. Otro elemento relevante es el incremento de la actividad glutamatérgica, que ha consolidado la indicación de bloqueadores del receptor de N-metil-D-aspartato como la memantina.
Tabla 3. Puntos fisiopatológicos clave de la enfermedad de Alzheimer y estrategias terapéuticas asociadas
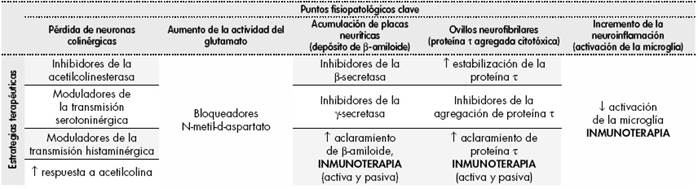
Ambos grupos terapéuticos son de uso corriente en la práctica clínica actual. Otros tres mecanismos fisiopatológicos pueden abrir nuevas dianas terapéuticas, esta vez por vía del uso de inmunoterapias. La acumulación de placas neuríticas (β-amiloide), de ovillos neurofibrilares (proteína τ) y la inflamación local promovida por la microglía, son potenciales objetivos terapéuticos que se están evaluando en los últimos años55.
El β-amiloide se forma en dos pasos desde la proteína precursora de amiloide (APP en su acrónimo en inglés), por vía de los complejos enzimáticos de la β-secretasa y de la γ - secretasa. Se piensa que el depósito de β -amiloide tendría un importante papel en el desarrollo de la enfermedad de Alzheimer56. Con esta hipótesis en mente, se han desarrollado, por un lado, terapias que puedan reducir la actividad de los complejos β-secretasa y γ-secretasa, con el objetivo de producir menos β -amiloide y, por otro, tratamientos inmunológicos que puedan aumentar la eliminación del β-amiloide patógeno ya formado.
Así, se han desarrollado dos estrategias, la inmunoterapia activa y la inmunoterapia pasiva. Uno de los primeros ensayos clínicos de inmunoterapia activa se realizó con AN1792, un péptido amiloide sintético (Aβ42) que induce la producción de anticuerpos contra el β-amiloide57. El ensayo clínico fase 2 fue detenido por la aparición de meningoencefalitis en el 6% de los sujetos del estudio. Además, en los que no desarrollaron meningoencefalitis, a pesar de una clara reducción de los depósitos de placas seniles, no se demostró un retraso en la evolución del deterioro cognitivo. En vista de estos resultados, se argumentó que los pacientes incluidos en el estudio fueron pacientes con enfermedad de Alzheimer moderada/grave, por lo que, quizás, el tratamiento con fármacos que promueven la eliminación de placas seniles deba ser indicado en fases más precoces de la patología. Con esta recomendación, se están evaluando otras moléculas que promueven una inmunidad activa para eliminar las placas amiloides. Entre ellas, el agente CAD106, seguro y bien tolerado, sin datos de meningoencefalitis y que se mantiene en estudio58. En cuanto a la inmunoterapia pasiva, se han desarrollado varias moléculas que están siendo evaluadas. Bapineuzumab, cuya diana es el extremo N-terminal del β-amiloide, ha sido abandonado por su escasa eficacia y negativo perfil de seguridad en fase 359,60. Solanezumab, cuyo objetivo es el β-amiloide monomérico soluble, sí que ha presentado un adecuado perfil de seguridad y se está evaluando en pacientes con formas muy precoces de la enfermedad de Alzheimer. Ha demostrado un aumento de proteína β-amiloide-42 soluble en el líquido cefalorraquídeo de los pacientes, efecto dosis -dependiente, lo que avalaría su mecanismo de acción de eliminación de los depósitos de β-amiloide. Sin embargo, no ha demostrado eficacia clínica en la enfermedad de Alzheimer leve61. Aducanumab también se encuentra en fase 3 de ensayos clínicos, siendo utilizado en la enfermedad de Alzheimer leve pero también en fase de deterioro cognitivo leve. Finalmente, gantenerumab, cuya función es interactuar con las fibrillas β-amiloides para reclutar microglía, activar la fagocitosis y degradar las placas neuríticas, está en fases precoces del desarrollo clínico, con un adecuado perfil de seguridad, y pendientes de resultados de eficacia en la enfermedad de Alzheimer leve y el deterioro cognitivo leve62.
De la misma forma que con las placas neuríticas, la presencia de ovillos neurofibrilares, formados por proteína τ hiperfosforilada, parece ser uno de los elementos clave responsables de la patogenia en la enfermedad de Alzheimer. La proteína τ agregada es citotóxica, por lo que evitar su producción o favorecer su eliminación podría tener un efecto clínico sobre los pacientes. Tal y como se ha comentado para la proteína β-amiloide, existen moléculas que, por vía de la inmunoterapia activa, podrían favorecer la eliminación de la proteína τ patogénica. AADvac -1, una vacuna activa con una forma natural truncada de τ, se encuentra en fase 2 tras haber mostrado un buen perfil de seguridad. C2N8E12 es un anticuerpo humanizado anti-τ que está actualmente en fase 2 y se esperan resultados de seguridad y eficacia para 202063.
Además de las placas neuríticas (β -amiloide) y de los ovillos neurofibrilares (τ), la neuroinflamación es uno de los puntos clave de la fisiopatología de la enfermedad de Alzheimer. Se ha evidenciado una clara astrogliosis, entre otros signos de inflamación, alrededor de las placas de amiloide y varios estudios sugieren una relación entre la activación de la microglía, la formación de placas neuríticas y la progresión clínica de la enfermedad. Por este motivo, una terapia dirigida a inhibir la activación de la microglía podría ser de utilidad. Sin embargo, resultados negativos con tramiprosato (falta de eficacia en fase 3), ibuprofeno y r-flurbiprofeno han reducido las expectativas en este grupo terapéutico/mecanismo de acción. En ensayo clínico fase 2 permanece CHF 5074, un modulador de microglía, para pacientes con deterioro cognitivo leve63.
Por último, cabe señalar que la inmunoglobulina humana intravenosa parece ser eficaz en el mantenimiento de la capacidad cognitiva de los pacientes con enfermedad de Alzheimer leve a moderada en ensayos clínicos de fase 364.
Sistema nervioso periférico
-
- SGB: se trata de una polirradiculopatía sensitivo-motora aguda, desmielinizante, axonal o mixta, de origen inflamatorio. El cuadro típico es de parestesias, dolor y pérdida de fuerza, aunque existen diferentes variantes clínicas, como el síndrome de Miller Fisher, un cuadro de ataxia sensitiva con afectación del tronco encefálico y los nervios oculomotores. La incidencia es de 0,4 a 3,25 pacientes por 100.000 habitantes y año65.
El tratamiento del SGB se realiza con plasmaféresis, cuya efectividad se confirma en la revisión de la Cochrane de 201266. Una alternativa es el uso de inmunoglobulina humana intravenosa 0,4 g/kg de peso/ día durante 5 días. No se han realizado estudios controlados con placebo, pero una revisión de la Cochrane de 2014 confirma su eficacia, comparable a la de la plasmaféresis tras una evaluación sistemática de cinco ensayos clínicos67. Además, dos ensayos clínicos confirman una similar eficacia pero menos efectos secundarios de las inmunoglobulinas con respecto a la plasmaféresis68,69. Una completa revisión sobre el tema puede ser consultada en el trabajo de Wijdicks y Klein70.
-
- Polineuropatía inflamatoria desmielinizante crónica (CIDP en su acrónimo en inglés): considerada la forma crónica (>8 semanas de duración) de la variante desmielinizante del SGB, su tratamiento consiste también en el uso de plasmaféresis o inmunoglobulinas. Sin embargo, mientras que en el SGB los glucocorticoides se consideran ineficaces, sí que son útiles tanto en la fase aguda como en el tratamiento de mantenimiento de la CIDP71. Rituximab ha mostrado eficacia en estudios de pequeñas cohortes de pacientes con esta patología72,73. Eculizumab podría ser una opción todavía no testada en la CIDP. Fingolimod74y alemtuzumab75podrían tener su papel en el tratamiento de la enfermedad, pendiente de confirmación en ensayos clínicos aleatorizados y controlados.
- Neuropatía motora multifocal (NMM): trastorno autoinmune de escasa prevalencia (0,6-2 pacientes por cada 100.000 habitantes)76. Produce una pérdida de fuerza lentamente progresiva, asimétrica y de predominio distal. Mediada por anticuerpos antigangliósidos, la NMM puede ser tratada con inmunoglobulina humana, tanto intravenosa como subcutánea, mejorando así los síntomas y evitando la progresión de los mismos. Cuatro ensayos clínicos77 78 79-80han demostrado que el 78% de los pacientes tratados con inmunoglobulinas intravenosas mejoraron significativamente su capacidad motora, comparados con un 4% que lo hicieron con placebo. A pesar de que el metaanálisis de dichos estudios no mostró diferencias significativas en la mejora de la discapacidad81, la European Federation of Neurological Societies (EFNS) recomienda en su guía de tratamiento el uso de 2 g/kg de peso de inmunoglobulina humana intravenosa para el tratamiento de primera línea de la NMM,
dosis que se administra a lo largo de dos a cinco días. La guía también indica que la dosis de mantenimiento de inmunoglobulina humana intravenosa que debe ser administrada tras la mejora inicial con el primer ciclo será de 1 g/kg de peso cada dos a cuatro semanas o 2 g/kg cada uno o dos meses82. Cabe señalar que la formulación subcutánea de inmunoglobulina ha demostrado eficacia superponible a la intravenosa en el tratamiento de la NMM, tanto para la fase inicial83, como para el mantenimiento84. Para una extensa revisión a este respecto, se recomienda el trabajo de Kumar y cols.85.
- Esclerosis lateral amiotrófica: es una enfermedad degenerativa de la motoneurona. Recientemente se han imputado mecanismos fisiopatológicos de sustrato inmunológico, abriéndose así una vía potencial para un tratamiento inmunoterápico. Una revisión sobre este tema86describe cómo se han utilizado tratamientos para la artritis reumatoide, anakinra (análogo recombinante del antagonista del receptor de la interleucina-1) con resultados negativos, mastinib (inhibidor de la tirosinacinasa) cuyo ensayo fase 3 está en curso, y tocilizumab, también en fase de ensayo clínico, en este caso fase 2. También se ha recurrido a tratamientos para la EM tales como acetato de glatiramero (resultados negativos) o fingolimod (eficacia no demostrada). Los resultados de ineficacia obtenidos con inmunoglobulinas intravenosas, celecoxib, ozanezumab, NP001 (taurina), talidomida, factor estimulador de crecimiento de granulocitos, ciclosporina o radiación linfoide total no han evitado que se continúe la línea de investigación de las terapias inmunológicas en la esclerosis lateral amiotrófica. Otros agentes, como el ibudilast (inhibidor de TLR4 y fosfodiesterasas 3 y 4), el RNS60 o fármacos utilizados para evitar el rechazo en los trasplantes (basiliximab + micofenolato mofetilo + tacrolimus + glucocorticoides) están actualmente en estudio.
- Miastenia gravis: se trata de una enfermedad mediada por anticuerpos (antirreceptor de acetilcolina (AChR) o anticinasa musculoespecífica (MuSK)) que impide la adecuada transmisión en la placa motora. Su síntoma característico es la fatigabilidad muscular. El tratamiento inicial, aparte de los inhibidores de la acetilcolinesterasa, se basa en el empleo de prednisona oral, inmunoglobulinas intravenosas y/o plasmaféresis para las recaídas de la enfermedad. Para evitar el uso continuado de glucocorticoides en pacientes con enfermedades generalizadas se utilizan azatioprina, micofenolato mofetilo, ciclosporina A, metotrexato, tacrolimus o ciclofosfamida. Para una completa revisión sobre el empleo de estos medicamentos en la miastenia gravis, se recomienda el trabajo de Lee y Jander87. Otra opción terapéutica es el rituximab, anticuerpo monoclonal quimérico anti-CD20, el cual se recomienda en pacientes con formas moderadas-graves de la enfermedad que son refractarios a otros tratamientos, y en aquéllos que son anti-MuSK positivos88,89. Un metaanálisis que evalúa 15 estudios clínicos no controlados con un total de 168 pacientes incluidos, con diferentes pautas de tratamiento con rituximab, parece mostrar su eficacia en el tratamiento de la miastenia gravis AchR positiva, MuSK positiva y AchR/MuSK doble negativa90. Más recientemente, eculizumab ha demostrado eficacia en un ensayo de fase 291y en otro de fase 392, motivo por el cual se podría plantear inicialmente como opción terapéutica en pacientes graves y refractarios a otras estrategias terapéuticas93.
- Miopatías autoinmunes: la prevalencia de la polimiositis y de la dermatomiositis, las dos miopatías autoinmunes más frecuentes, es de 21,5/100.000 habitantes94. El tratamiento de ambas y de la miopatía necrotizante inmunomediada es, en primera línea, con corticoesteroides95 96-97. En los pacientes con clínica grave (disfagia o incapacidad para caminar) se utiliza metilprednisolona intravenosa 1 g/día/3 días, seguida de prednisona oral en pauta descendente desde una dosis de 60 mg/día de prednisona. En casos moderados se puede empezar con la pauta oral sin la dosis de carga intravenosa previa. En presentaciones clínicas más leves se puede comenzar con dosis más bajas de prednisona. Una vez que la fuerza muscular se normaliza, se comienza la reducción progresiva de dosis. En pacientes con enfermedad grave, en aquéllos con respuesta incompleta a corticoides tras dos meses en tratamiento y en aquéllos en los que no se puede reducir la dosis de éstos por debajo de 10 mg/día, se recomienda el uso de azatioprina, metotrexato o micofenolato mofetilo. Si existe fracaso o intolerancia de estos medicamentos de segunda línea, se podría recurrir a rituximab, ciclosporina, ciclofosfamida o bloqueadores de TNFα98. La inmunoglobulina humana intravenosa es eficaz en el tratamiento de la dermatomiositis99y probablemente también en la polimiositis. Se puede usar en segunda línea en pacientes con clínica grave, dado que su efecto es más rápido que los medicamentos de dicha línea terapéutica. La dosis es la habitual de 2 g/kg de peso, distribuida en tres a cinco días. La miositis por cuerpos de inclusión no responde a la inmunoterapia.
Discusión
En la actualidad, los tratamientos inmunológicos nos permiten tratar numerosas patologías neurológicas de forma más precisa e individualizada. Productos diseñados con dianas terapéuticas cada vez más específicas y evaluados mediante ensayos clínicos con alto nivel de evidencia están apareciendo con mayor frecuencia en la práctica clínica. Además, el espectro de enfermedades potencialmente tratables mediante inmunoterapia es cada vez mayor y comienza a incluir patologías neurológicas de alta prevalencia como la migraña y, posiblemente, la enfermedad de Alzheimer. El alto impacto sociosanitario que dichas terapias pueden traer aparejado obligará a compromisos y consensos por parte de todos los actores implicados en su adecuada utilización.
REFERENCIAS
Otero-Romero S, Roura P, Solà J, Altimiras J, Sastre-Garriga J, Nos C, et al. Increase in the prevalence of multiple sclerosis over a 17-year period in Osona, Catalonia, Spain. Mult Scler. 2013;19(2):245-8. [ Links ]
Le Page E, Veillard D, Laplaud DA, Hamonic S, Wardi R, Lebrun C, et al. Oral versus intravenous high-dose methylprednisolone for treatment of relapses in patients with multiple sclerosis (COPOUSEP): a randomised, controlled, double-blind, noninferiority trial. Lancet. 2015;386(9997):974-81. [ Links ]
Ehler J, Koball S, Sauer M, Mitzner S, Hickstein H, Benecke R, et al. Response to therapeutic plasma exchange as a rescue treatment in clinically isolated syndromes and acute worsening of multiple sclerosis: a retrospective analysis of 90 patients. PLoS One. 2015;10(8):e0134583. [ Links ]
Miller AE. Oral teriflunomide in the treatment of relapsing forms of multiple sclerosis: clinical evidence and long-term experience. Ther Adv Neurol Disord. 2017;10(12):381-96. [ Links ]
Linker RA, Haghikia A. Dimethyl fumarate in multiple sclerosis: latests developments, evidence and place in therapy. Ther Adv Chronic Dis. 2016;7(4):198-207. [ Links ]
Tramacere I, Del Giovane C, Salanti G, D’Amico R, Filippini G. Immunomodulators and immunosuppressants for relapsing-remitting multiple sclerosis: a network metaanalysis. Cochrane Database Syst Rev. Sep 18;(9):CD011381. [ Links ]
Zhang J, Shi S, Zhang Y, Luo J, Xia Y, Meng L, et al. Alemtuzumab versus interferon beta 1a for relapsing-remitting multiple sclerosis. Cochrane Database Syst Rev. Nov 27;(11):CD010968. [ Links ]
Chaundry BZ, Cohen JA, Conway DS. Sphingosine 1-Phosphate receptor modulators for the treatment of multiple sclerosis. Neurotherapeutics. 2017;14(4):859-73. [ Links ]
Kappos L, Bar-Or A, Cree BAC, Fox RJ, Giovannoni G, Gold R, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a doubleblind, randomised, phase 3 study. Lancet. 2018;391(10127):1263-73. [ Links ]
Gelfand JM, Cree BAC, Hauser SL. Ocrelizumab and other CD20+ B-cell-depleting therapies in multiple sclerosis. Neurotherapeutics. 2017;14(4):835-41. [ Links ]
Giovannoni G. Cladribine to treat relapsing forms of multiple sclerosis. Neurotherapeutics. 2017;14(4):874-87. [ Links ]
Weinshenker BG, O´Brien PC, Petterson TM, Nosewortht JH, Lucchinetti CF, Dodick DW, et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol. 1999;46(6):878-86. [ Links ]
Elsone L, Panicker J, Mutch K, Boggild M, Appleton R, Jacob A. Role of intravenous immunoglobulin in the treatment of acute relapses of neuromyelitis optica: experience in 10 patients. Mult Scler. 2014;20(4):501-4. [ Links ]
Flanagan EP. Autoimmune myelopathies. Handb Clin Neurol. 2016;133:327-51. [ Links ]
Kitley J, Elsone L, George J, Waters P, Woodhall M, Vicent A, et al. Methotrexate is an alternative to azathioprine in neuromyelitis optica spectrum disorders with aquoporin-4 antibodies. J Neurol Neurosurg Psychiatry. 2013;84(8):918-21. [ Links ]
Flanagan EP, Weinshenker BG. Neuromyelitis spectrum disorders. Curr Neurol Neurosci Rep. 2014;14(9):483. [ Links ]
Palace J, Leite MI, Nairne A, Vincent A. Interferon beta treatment in neuromyelitis optica: increase in relapses and aquoporin 4 antibody titers. Arch Neurol. 2010;67(8):1016-7. [ Links ]
Jacob A, Hutchinson M, Elsone L, Kelly S, Ali R, Saulans I, et al. Does natalizumab therapy worsen neuromyelitis optica? Neurology. 2012;79(10):1065-6. [ Links ]
Min JH, Kin BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler. 2012;18(1):113-5. [ Links ]
Kieseier BC, Stuve O, Dehmel T, Goebels N, Leussink VI, Mausberg AK, et al. Disease amelioration with tocilizumab in a treatment-resistant patient with neuromyelitis optica: implication for cellular immune responses. JAMA Neurol. 2013;70(3):390-3. [ Links ]
Ayzenberg I, Kleiter I, Schröder A, Hellwig K, Chan A, Yamamura T, et al. Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-CD20 therapy. JAMA Neurol. 2013;70(3):394-7. [ Links ]
Araki M, Aranami T, Matsuoka T, Nakamura M, Miyake S, Yamamura T. Clinical improvement in a patient with neuromyelitis optica following therapy with the anti-IL-6 receptor monoclonal antibody tocilizumab. Mod Rheumatol. 2013;23(4):827-31. [ Links ]
Harmel J, Rigelstein M, Ingwersen J, Mathys C, Goebels N, Hartung HP, et al. Interferon-β-related tumefactive brain lesion in a Causcasian patient with neuromyelitis optica and clinical stabilization with tocilizumab. BMC Neurol. 2014;14:247. [ Links ]
Komai T, Shoda H, Yamaguchi K, Sakurai K, Shibuya M, Kubo K, et al. Neuromyelitis optica spectrum disorder complicated with Sjögren syndrome successfully treated with tocilizumab: a case report. Mod Rheumatol. 2016;26(2):294-6. [ Links ]
Ringelstein M, Ayzenberg I, Harmel J, Lauenstein AS, Lensch E, Stögbauer F, et al. Long-term therapy with interleukin 6 receptor blockade in highly active neuromyelitis optica spectrum disorder. JAMA Neurol. 2015;72(7):756- 63. [ Links ]
Pittock SJ, Lennon VA, McKeon A, Mandrekar J, Weinshenker BG, Lucchinetti GF, et al. Eculizumab in AQP4- IgG-positive relasing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. 2013;12(6):554-62. [ Links ]
Lin J, Xue B, Li X, Xia J. Monoclonal antibody therapy for neuromyelitis optica spectrum disorder: current and future. Int J Neurosci. 2017;127(8):735-44. [ Links ]
Steiner TJ, Stovner LJ, Birbeck GL. Migraine: the seventh disabler. J Headache Pain. 2013;14:1. [ Links ]
Silberstein SD, Holland S, Freitag F, Dodick DW, Argoff C, Ashman E; Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Evidence-based guideline update: pharmacologic treatment for episodic migraine prevention in adults. Neurology. 2012;78(17):1337-45. [ Links ]
Aurora SK, Dodick DW, Turkel CC, DeGryse RE, Silberstein SD, Lipton RB, et al.; PREEMPT 1 Chronic Migraine Study Group. OnabotulinumtoxinA for treatment of chronic migraine: Results from the double-blind, randomized, placebo-controlled phase of the preempt 1 trial. Cephalalgia. 2010;30(7):793-803. [ Links ]
Diener HC, Dodick DW, Aurora SK, Turkel CC, DeGryse RE, Lipton RB, et al.; PREEMPT 1 Chronic Migraine Study Group OnabotulinumtoxinA for treatment of chronic migraine: Results from the double-blind, randomized, placebo-controlled phase of the preempt 2 trial. Cephalalgia. 2010;30(7):804-4. [ Links ]
Dodick DW, Turkel CC, DeGryse RE, Aurora SK, Silberstein SD, Lipton RB, et al.; PREEMPT 1 Chronic Migraine Study Group OnabotulinumtoxinA for treatment of chronic migraine: Pooled results from the double-blind, randomized, placebo-controlled phases of the preempt clinical program. Headache. 2010;50(6):921-36. [ Links ]
Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol. 1993;33(1):48-56. [ Links ]
Lassen LH, Haderslev PA, Jacobsen VB, Iversen HK, Sperling B, Olesen J. CGRP may play a causative role in migraine. Cephalalgia. 2002;22(1):54-61. [ Links ]
Dodick DW, Goadsby PJ, Silberstein SD, Lipton RB, Olesen J, Ashina M, et al. Safety and efficacy of ALD403, an antibody to calcitonin gene-related peptide, for the prevention of frequent episodic migraine: a randomized, double- blind, placebocontrolled, exploratory phase 2 trial. Lancet Neurol. 2014;13(11):1100-7. [ Links ]
Dodick DW, Goadsby PJ, Spierings EL, Scherer JC, Sweeney SP, Grayzel DS. Safety and efficacy of LY2951742, a monoclonal antibody to calcitonin gene-related peptide, for the prevention of migraine: a phase 2, randomized, double-blind, placebo-controlled study. Lancet Neurol. 2014;13(9):885-92. [ Links ]
Bigal ME, Dodick DW, Rapoport AM, Silberstein SD, Ma Y, Yang R, et al. Safety, tolerability and efficacy of TEV- 48125 for preventive treatment of high-frequency episodic migraine: a multicenter, randomized, double-blind, placebo- controlled, phase 2b study. Lancet Neurol. 2015;14(11):1081-90. [ Links ]
Bigal ME, Edvinsson L, Rapoport AM, Lipton RB, Spierings EL, Diener HC, et al. Safety, tolerability and efficacy of TEV-48125 for preventive treatment of chronic migraine: a multicenter, randomized, double-blind, placebo-controlled, phase 2b study. Lancet Neurol. 2015;14(11):1091-100. [ Links ]
Sun H, Dodick DW, Silberstein S, Goadsby PJ, Reuter U, Ashina M, et al. Safety and efficacy of AMG 334 for prevention of episodic migraine: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15(4):382-90. [ Links ]
Tepper S, Ashina M, Reuter U, Brandes JL, Dolezil D, Silberstein S, et al. Safety and efficacy of erenumab for preventive treatment of chronic migraine: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017;16(6):425-34. [ Links ]
Deen M, Correnti E, Kamm K, Kelderman T, Papetti L, Rubio-Beltrán E, et al. Blocking CGRP in migraine patients - a review of pros and cons. J Headache Pain. 2017;18(1):96. [ Links ]
McKeon A, Vincent A. Autoimmune movement disorders. Handb Clin Neurol. 2016;133:301-15. [ Links ]
Mohammad SS, Dale RC. Principles and approaches to the treatment of immunemediated movement disorders. Eur J Paediatr Neurol. 2018;22(2):292-300. [ Links ]
Tysnes OB, Storstein A. Epidemiology of Parkinson´s disease. J Neural Transm (Vienna). 2017;124(8):901-5. [ Links ]
Oertel W, Schultz JB. Current and experimental treatments of Parkinson disease: a guide for neuroscientists. J Neurochem. 2016;139(Suppl 1):325-37. [ Links ]
Brundin P, Dave KD, Kordower JH. Therapeutic approaches to target alpha-synuclein pathology. Exp Neurol. 2017;298(Pt B):225-35. [ Links ]
Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404. [ Links ]
Dalmau J, Lancaster E, Martínez-Hernández E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10(1):63-74. [ Links ]
Suleiman J, Dale RC. The recognition and treatment of autoimmune epilepsy in children. Dev Med Child Neurol. 2015;57(5):431-40. [ Links ]
Irani SR, Gelfand JM, Bettcher BM, Singhal NS, Geschwind MD. Effect of rituximab in patients with leucine-rich, glioma-inactivated 1 antibody-associated encephalopathy. JAMA Neurol. 2014;71(7):896-900. [ Links ]
Flanagan EP, Drubach DA, Boeve BF. Autoimmune dementia and encephalopathy. Handb Clin Neurol. 2016;133:247-67. [ Links ]
Pittock SJ, Palace J. Paraneoplastic and idiopathic autoimmune neurologic disorders: approach to diagnosis and treatment. Handb Clin Neurol. 2016;133:165-83. [ Links ]
Pohl D, Alper G, van Haren K, Konberg AJ, Lucchinetti CF, Tenembaum S, et al. Acute disseminated encephalomyelitis: updates on an inflammatory CNS syndrome. Neurology. 2016;87(9 Suppl 2):S38-45. [ Links ]
Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology. 2013;80(19):1778-83. [ Links ]
Hung SY, Fu WM. Drug candidates in clinical trials for Alzheimer´s disease. J Biomed Sci. 2017;24(1):47. [ Links ]
Graham WV, Bonito-Oliva A, Sakmar TP. Update on Alzheimer´s disease therapy and prevention strategies. Annu Rev Med. 2017;68:413-30. [ Links ]
Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64(9):1553-62. [ Links ]
Farlow MR, Andreasen N, Riviere ME, Vostiar I, Vitaliti A, Sovago J, et al. Long-term treatment with active Aβ immunotherapy with CAD106 in mild Alzheimer´s disease. Alzheimer´s Res Ther. 2015;7(1):23. [ Links ]
Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Azheimer´s disease. N Engl J Med. 2014;370(4):322-33. [ Links ]
Vandenberghe R, Rinne JO, Boada M, Katayama S, Scheltens P, Vellas B, et al. Bapineuzumab for mild to moderate Alzheimer´s disease in two global, randomized, phase 3 trials. Alzheimer´s Res Ther. 2016;8(1):18. [ Links ]
Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, et al. Trial of Solanezumab for mild dementia due to Alzheimer´s disease. N Engl J Med. 2018;378(4):321-30. [ Links ]
Mo JJ, Li JY, Yang Z, Liu Z, Feng JS. Efficacy and safety of anti-amyloid-β immunotherapy for Alzheimer’s disease: a systematic review and network meta-analysis. Ann Clin Transl Neurol. 2017;4(12):931-42. [ Links ]
Bittar A, Sengupta U, Kayed R. Prospects for strain-specific immunotherapy in Alzheimer´s disease and taupathies. NPJ Vaccines. 2018;3:9. [ Links ]
Relkin NR, Thomas RG, Rissman RA, Brewer JB, Rafii MS, van Dyck CH, et al. A phase 3 trial of IV immunoglobulin for Alzheimer disease. Neurology. 2017;88(18):1768-75. [ Links ]
Wilson HJ, Goodfellow JA. GBS100: celebrating a century of progress in Guillain-Barré syndrome. La Jolla, CA: Peripheral Nerve Society; 2016. [ Links ]
Raphaël JC, Chevret S, Hughes RA, Annane D. Plasma exchange for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2012;7:CD001798. [ Links ]
Hughes RA, Swan AV, van Doom PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2014;9:CD002063. [ Links ]
Van der Meché FG, Schmidt PI, Dutch Guillain-Barré Study Group. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. N Engl J Med. 1992;326(17):1123-9. [ Links ]
Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group. Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barré syndrome. Lancet. 1997;349(9047):225-30. [ Links ]
Wijdicks EFM, Klein CJ. Guillain-Barré syndrome. Mayo Clin Proc. 2017;92(3):467-79. [ Links ]
Hu MY, Stathopoulos P, O´Connor KC, Pittock SJ, Nowak RJ. Current and future immunotherapy targets in autoimmune neurology. Handb Clin Neurol. 2016;132:511-36. [ Links ]
Benedetti L, Briani C, Franciotta D, Fazio R, Paolasso I, Comi C, et al. Rituximab in patients with chronic inflammatory demyelinating polyradiculoneuropathy: a report of 13 cases and review of the literature. J Neurol Neurosurg Psychiatry. 2011;82(3):306-8. [ Links ]
D´Amico A, Catteruccia M, De Benedetti F, Vivarelli M, Colucci M, Cascioli S, et al. Rituximab in childhood onset idiopathic refractory chronic inflammatory demyelinating polyneuropathy. Eur J Pediatr Neurol. 2012;16(3):301-3. [ Links ]
Zhang Z, Zhang ZY, Fauser U, Schluesener HJ. FTY720 ameliorates experimental autoinmune neuritis by inhibition of lymphocyte and monocyte infiltration into peripheral nerves. Exp Neurol. 2008;210(2):681-90. [ Links ]
Marsh EA, Hirst CL, Llewelyn JG, Cossburn MD, Reilly MM, Krishnan A, et al. Alemtuzumab in the treatment of IVIG-dependent chronic inflammatory demyelinating polyneuropathy. J Neurol. 2010;257(6):913-9. [ Links ]
Nobile-Ozario E. Multifocal motor neuropathy. J Neuroimmunol. 2001;115(1-2):4-18. [ Links ]
Azulay JP, Blin O, Puget J, Boucraut J, Billé-Turc F, Carles G, et al. Intravenous immunoglobulin treatment in patients with motor neuron syndromes associated with anti-GM1 antibodies: a double-blind, placebo-controlled study. Neurology. 1994;44(3Pt 1):429-32. [ Links ]
Van den Berg LH, Kerkhoff H, Oey PL, Franssen H, Mollee I, Vermeulen M, et al. Treatment of multifocal motor neuropathy with high dose intravenous immunoglobulins: a double-blind, placebo controlled study. J Neurol Neurosurg Psychiatry. 1995;59(3):248-52. [ Links ]
Federico P, Zochodone DW, Hahn AF, Brown WF, Feasby TE. Multifocal motor neuropathy improved by IVIg: randomized, double-blind, placebo-controlled study. Neurology. 2000;55(9):1256-62. [ Links ]
Leger JM, Chassande B, Musset L, Meininger V, Bouche P, Baumann N. Intravenous immunoglobulin therapy in multifocal motor neuropathy: a double-blind, placebocontrolled study. Brain. 2001;124(Pt 1):145-53. [ Links ]
Van Shaik IN, van den Berg LH, de Haan R, Vermeulen M. Intravenous immunoglobulin treatment for multifocal motor neuropathy. Cochrane Database Syst Rev. April 18 (2):CD004429. [ Links ]
Joint task force of the EFNS ang the PNS. EFNS/PNS guidelines on management of multifocal motor neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society - first revision. J Peripher Nerv Syst. 2010;15(4):295-301. [ Links ]
Harbo T, Andersen H, Hess A, Hansen K, Sindrup SH, Jakobsen J. Subcutaneous versus intravenous immunoglobulin in mulifocal motor neuropathy: a randomized single-blinded cross-over trial. Eur J Neurol. 2009;16(5):631-8. [ Links ]
Harbo T, Andersen H, Jakobsen J. Long-term therapy with high doses of subcutaneous immunoglobulin in multifocal motor neuropathy. Neurology. 2010;75(15):1377-80. [ Links ]
Kumar A, Patwa HS, Nowak RJ. Immunoglobulin therapy in the treatment of multifocal motor neuropathy. J Neurol Sci. 2017; 375:190-7. [ Links ]
Khalid SI, Ample L, Kely R, Ladha SS, Dardis C. Immune modulation in the treatment of amyotrophic lateral sclerosis: a review of clinical trials. Front Neurol. 2017;8:486. [ Links ]
Lee JI, Jander S. Myasthenia gravis: recent advances in immunopathology and therapy. Expert Rev Neurother. 2017;17(3):287-99. [ Links ]
Díaz-Manera J, Martínez-Hernández E, Querol L, Klooster R, Rojas-García R, Suárez-Calvet X, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology. 2012;78(3):189-93. [ Links ]
Keung B, Robeson KR, DiCapua DB, Rosen JB, O´Connor KC, Goldstein JM, et al. Long-term benefit of rituximab in MuSK autoantibody myasthenia gravis patients. J Neurol Neurosurg Psychiatry. 2013;84(12):1407-9. [ Links ]
Iorio R, Damato V, Alboini PE, Evoli A. Efficacy and safety of rituximab for myasthenia gravis: a systematic review and meta-analysis. J Neurol. 2015;262(5):1115-9. [ Links ]
Howard JF, Barohn RJ, Cutter GR, Freimer M, Juel VC, Mozaffar T, et al. A randomized, double-blind, placebo- controlled phase II study of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve. 2013;48(1):76-84. [ Links ]
Howard JF, Utsugisawa K, Benatar M, Murai H, Barohn RJ, Illa I, et al. Safety and efficacy of eculizumab in anti- acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. 2017;16(12):976-86. [ Links ]
Dhillon S. Eculizumab: a review in generalized myasthenia gravis. Drugs. 2018;78(3):367-76. [ Links ]
Bernatsky S, Josep L, Pineau CA, Bélisle P, Boivin JF, Banerjee D, et al. Estimating the prevalence of polymyositis and dermatomyositis from administrative data: age, sex and regional differences. Ann Rheum Dis. 2009;68(7):1192-6. [ Links ]
Amato AA, Russell JA. Inflammatory myopathies. In: Neuromuscular disorders. NewYork: McGraw Hill; 2008; p. 681. [ Links ]
Amato AA, Barohn RJ. Evaluation and treatment of inflammatory neuropathies. J Neurol Neurosurg Psychiatry. 2009;80(10):1060-8. [ Links ]
Gordon PA, Winer JB, Hoogendijk JE, Choy EH. Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database Syst Rev. Aug 15;(8):CD003643. [ Links ]
Mammen A. Autoimmune muscle disease. Handb Clin Neurol. 2016;133:467-84. [ Links ]
Dalakas MC, Illa I, Dambrosia JM, Soueidan SA, Stein DP, Otero C, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment of dermatomyositis. N Engl J Med. 1993;329(27):1993-2000. [ Links ]
Recibido: 05 de Abril de 2018; Aprobado: 30 de Julio de 2018
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
