










 Servicios personalizados
Servicios personalizados
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroduction
According to the report “Health at a Glance: Europe 2016”1, population aging and longer life expectancy have increased the burden of healthcare systems. This healthcare rising cost is a serious issue for most EU countries, which maintain an almost universal health coverage1,2. Accordingly, the incorporation of therapeutic innovations -which are increasingly expensive- to the benefits guaranteed by the National Health System must fulfill a double objective: guaranteeing patients' access to innovative medicines, without undermining the system's economic sustainability. Reaching a necessary balance between this double objective and the uncertainties generated by the incorporation of some medicines have led to the development of innovative experiences for its payment that result in new relationship scenarios between those responsible for the purchase and medicine suppliers. These type of agreements are known as risk-sharing agreements (RSA) and broadly apply to all relationship and contracting schemes that link the price of a therapeutic innovation with a series of variables related to objectives and results.
These financing schemes are not new in the field of medicines. Over the past 25 years, there have been multiple examples, as shown Carlson et al. review, in which 437 RSAs were identified worldwide3. The pace of adoption of these agreements has varied by country, the most active being Australia, Italy, Sweden, the United States and the United Kingdom, in which an upward trend has been renewed after a break in 2012-2013. The RSA typology observed covers a wide gradient that goes from the simplest forms of price-volume agreements to the most elaborate forms of contracts on effectiveness with guaranteed results. The latter are the ones that draws the most interest today because the resources investment is justified when translated into a real benefit in clinical practice conditions.
In Spain, RSA experiences are scarce and little information is available. The first RSA based on clinical results took place in February 2011 at hospital level (Virgen de las Nieves Hospital, Granada) for ambrisentan acquisition for treating pulmonary hypertension. That same year, the first RSA was signed between a Public Administration and a pharmaceutical company, specifically between Institut Català d'Oncologia, Servei Català de Salut (CatSalut) and the pharmaceutical laboratory that sold gefitinib for non-small cell lung cancer. This pilot agreement, whose results have been published recently4, has allowed the implementation of other payment schemes for results in CatSalut5 following a methodological guide with application criteria of RSA in the pharmacotherapy field6.
Orphan drugs (OD) are suitable candidates to be part of a payment program by clinical results. According to current European legislation, these are drugs indicated to treat rare diseases, which do not affect more than five people per 10,000 and lack alternative treatment7. The two characteristics that make these medications optimal for the application of a payment scheme linked to results are: 1) Its high economic impact, and 2) high uncertainty regarding efficacy and safety, as some are obtained through conditionally marketing authorization, and some under exceptional circumstances8.
Therefore, we consider the implementation of an RSA program in the acquisition of enzyme replacement therapies (ERT). They are indicated for a group of congenital metabolism errors caused by some of the lysosomal functions' deficit. There are more than 50 different clinical entities described, with the prevalence of 1/7,700 newborns9. ERT slows the progression of the disease and improves many of the clinical symptoms. However, due to its large size, it does not diffuse freely through the membranes nor reach therapeutic concentrations in some target tissues10.
The aim of this paper is to detail an implemented risk-sharing program's design and achievement for the ERT acquisition, as well as to show clinical and economic results derived from a referral hospital's program for congenital metabolic diseases.
Methods
A risk-sharing program was designed and implemented for the ERT acquisition in a hospital designated as a Reference Center, Service and Unit (RCSU) for congenital metabolic diseases, both for children and adults. The program began in January 2012 and all newly diagnosed patients with lysosomopathies and ERT prescribed were included. The work sequence that was carried out in the design of the program and the conclusion of agreements had the following phases:
Defining the response to treatment variables and criteria and establishing a treatment effectiveness gradation with the prescribing physician and the laboratory's medical department. Firstly, the efficacy and safety clinical variables of clinical trials and clinical guidelines were reviewed for each drug. The doctor was informed of the patient's incorporation into the program. An attempt was made to assemble the available bibliography in terms of response variables to the clinical reality of each patient. To facilitate the conclusion of the agreement's economic section, different steps of treatment effectiveness were established based on the number of clinical variables that the patient met (full, moderate, mild effectiveness, no response). Finally, the proposal prepared jointly between the doctor and the pharmacist was presented to the laboratory's medical department, and pertinent modifications were made until the three parties agreed.
In our hospital, the chosen risk-sharing scheme was the one that linked a percentage of reimbursement by the laboratory, previously agreed between the parties, to the effectiveness of the drug in actual practice. Thus, the Pharmacy Service and the laboratory's economic department met to assign said discount percentage to each level of effectiveness, where the hospital may proceed to a full payment whenever the effectiveness achieved was of 100%.
Preparation and signing of the agreement by all parties. Each agreement consisted of a common part constituted by the clauses that the legal cabinets considered appropriate: objective of the contract, description and characteristics, conditions of performance, participants, economic conditions, the center's obligations, informed consent, pharmacovigilance, duration, agreement resolution, confidentiality agreement, data protection and jurisdiction. The specific part for each patient was entitled Annex I, where the disease, the response variables and the effectiveness scale are described. Each document was signed by the hospital manager and the pharmaceutical company. In addition, Annex I required the signature of the responsible physician and the head of the Pharmacy Service.
Following the signing of the contract, the patient was informed of their inclusion in the program and therefore signed the informed consent and started the therapy at the day hospital. Implementing the agreement did not change the clinical practice regarding the prescribed dosage.
In order to know the exact number of vials that each of the patients consumed, a strict doses and vials control used for each patient was carried out, without modifying the usual work dynamics in which vials are shared for the patients with an appointment during the same day.
One year after the onset of the treatment, the pharmacist met with the doctor responsible for the patient and response variables were evaluated. It was determined whether the effectiveness of the treatment was full, moderate or mild. All this process was reflected in a report sent to the laboratory in order to apply the agreed discounts whenever effectiveness had not been fully achieved. The therapy's effectiveness evaluation -within the risk-sharing program- was carried out for two years in all patients except for two Hurler's disease patients who were evaluated for three years.
Results
Eight patients were included in the program (four with Hurler's disease, two with Pompe and two with Gaucher), five of them were women and three were men.
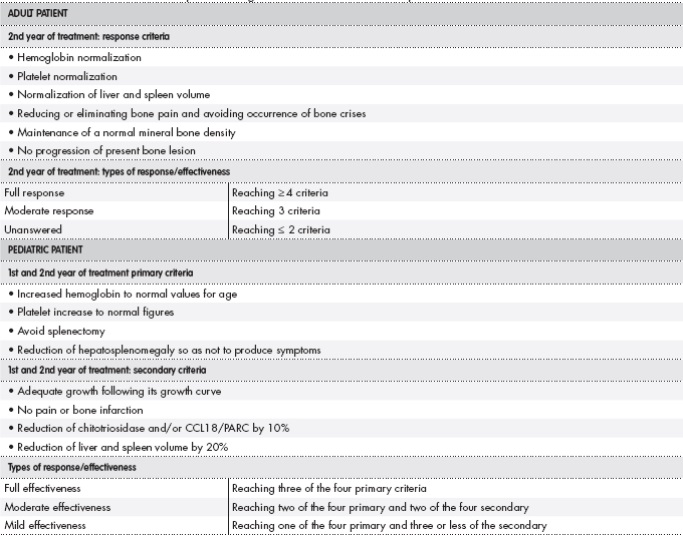
The effectiveness criteria and response types were defined for each ERT, depending on whether it was the first year of treatment or later and each patient's age. The four patients with Hurler's disease were children between one and three years old. Table 1 shows their defined clinical variables. For these patients, it was necessary to define effectiveness criteria and different response types after one and two years of treatment due to both the important clearance of glycosaminoglycan deposits (GAGs) that occurs during the first months of treatment, and the thereof levels stabilization that is observed in the second year, according to the experience of our doctors of the metabolic unit and the available evidence11,12. The two Pompe disease patients were adults (46 and 40 years old), so the definition of the response criteria was based on the clinical variables defined in the pivotal trials, which in turn were the same as evaluated by the doctors of our hospital in their clinical practice13 (Table 2). Finally, regarding the two Gaucher's disease patients, one was an adult (42 years old) and the other was pediatric (two years old). For this disease, the literature is more abundant and the clinical guidelines themselves define the treatment effectiveness criteria and types of response in both children and adults14,15. Thus, the adult patient, whose predominant symptoms were bone related, three of the six defined criteria were related to bone pathology. However, in the case of the affected infant, other primary and secondary criteria were more adjusted to the age of the patient, shown in Table 3.
Table 1. Efficacy criteria and response to laronidase in pediatric Hurler's disease patients

FCV: forced vital capacity; GAG: glycosaminoglycan.

Each patient was measured for clinical variables that were part of the response criteria before starting treatment and annually after ERT administration. The four Hurler patients (RSA1, RSA4, RSA6 and RSA9) showed a decrease of GAGs in urine for more than 40% during the first year of therapy with respect to baseline levels. This decrease continued for the four patients at the second year of treatment. The follow-up of RSA1 and RSA4 patients was maintained for a total period of three years (Figure 1). Both patients presented a stabilization -or even a small increase- in GAGs as was contemplated in the elaborated effectiveness criteria. As for the rest of the variables, respiratory tests were not performed by cause of impossibility of collaborating due to their young age. Liver and spleen sizes were normalized and doctors of different specialties confirmed the non-progression of the disease.

Figure 1. Evolution of glycosaminoglycans (GAGs) in Hurler's disease patients treated with laronidasa.
Of the two Pompe patients that were included in the program, patient RSA3 started treatment in July 2012 and had to be suspended due to concomitant oncological pathology. Although this patient resumed treatment months later, she was removed from the program since it was considered that the side effects of chemotherapy could dilute the effectiveness of ERT. On the other hand, patient RSA10 underwent the three tests defined in the agreement. The clinical examination with muscular strength evaluation after one year was comparable to that performed before treatment. After two years, some muscle groups were even improved in strength. In the six-minute walk test, the patient walked 318 meters (pre-treatment), 306 meters (at first year) and 341 meters (at two years). Respiratory function tests showed improvement after one and two years in some parameters, specifically regarding maximal inspiratory pressure (MIP) and maximal expiratory pressure (MEP). Specifically, MIP evolved from 54% pre- treatment, to 70% at first year and 58% at two years. As of MEP, from 47% pre-treatment to 63% at first year and 53% at two years. Therefore, RSA10 full effectiveness was achieved by not obtaining deterioration in any of the three evaluated parameters.
The effectiveness of adult patient Gaucher treatment was evaluated two years after the onset, as recommended by literature15. Infusions were able to normalize hemoglobin levels (12.2 g/dL), platelet count (179 thousand/mm3) and liver and spleen volumes. Regarding bone parameters, during therapy, the patient did not present bone pain, and the bone mineral density was normal -both in the lumbar spine and in the femoral head-. Bone lesions could not be assessed since the patient underwent MRI several months after the biennial evaluation. Anyway, the patient presented an optimal response, as five criteria of the established six were reached. As for the pediatric Gaucher's disease patient, the four primary criteria were met both after one and two years of the onset of therapy, and the four secondary criteria were all met except the decrease in chitotriosidase in the second year, which increased from 2,065 to 2,528 nmol/mL/h. However, the effectiveness was full when the four primary criteria were met.
In February 2016, the eight RSAs were completed. All were fully effective after two or three years of follow-up, except for the RSA3, which could not be evaluated. Given the effectiveness achieved, the hospital made full payment of all administered therapies.
Discussion
The implanted risk-sharing program is Spain's first published event of payment for clinical results using orphan drugs. Morel et al. reviewed risk-sharing agreements applied to ODs implemented by health authorities in seven European countries. Italy was the country with the highest number of schemes, and antineoplastic drugs were the most prevalent ODs in the agreements (50% financial agreements and 50% RSA based on clinical results)16. Several Italian publications list RSA-linked drugs based on clinical results. Among them are some ODs, which are either indicated in rare oncological diseases17,18 or idiopathic pulmonary fibrosis19. However, the information detailed is very limited. These publications only indicate the type of scheme applied to each drug without delving into their characteristics and description.
The implementation of the first RSAs was carried out in the hospitals. Negotiation processes first developed with price-volume agreements and later with evaluation of results agreements. The opacity of these decisions, which are common in Europe, has deprived us of learning in detail such events. In recent years, several European countries (United Kingdom, Italy, France, Germany and recently Eastern Europe) have been promoting national risk-sharing policies for the purchase of medicines, which has allowed a greater transparency20. However, in order to apply these policies at a national level, it is necessary, according to Gonçalves et al. to create and update computerized network records where the necessary data for the implementation of agreements based on clinical results are included21. This requirement, along with the complexity of the formulation and the high need for management required by these agreements, are the reasons why Spanish health authorities have ediscarded their use at a national level, positioning them at a regional or hospital level.
Specifically, in the course of the implementation of our agreements, we encountered inconveniences due to the limited experience that existed in this regard. 1) The industry was reluctant to assume the risk that the drug did not have the expected real effectiveness. Therefore, the applicable discount was higher than expected to maintain the medicine's sustainability in the market. 2) We found it especially difficult to define and agree with the clinicians and the laboratories' medical departments on the clinical variables and their measurement times. 3) The organization and monitoring of the RSAs were initially complex and required resources for their implementation and periodic evaluation. Accordingly, we intended to simplify the process and create a standard model of agreement that undoubtedly reduced the administrative burden. However, the strengthening of structures and information systems in the healthcare administration would facilitate the analysis and monitoring of these agreements, and would greatly improve monitoring from an operational point of view22.
The economic impact of the risk-sharing program implementation for our patients was very limited since the effectiveness of every case was fully achieved. However, the decision to use payment schemes by results was not based only on economic issues, but on dispelling the uncertainties of the effectiveness of the treatments. Following this decision, Clopés et al. described a discrete economic impact, saving 4.5% of the total cost of gefitinib4. Authors noted that the potential impact of payment agreements by results limiting the conditions of use of the therapy was much more economically relevant than the savings themselves. Thus, we believe that the greatest achievement reached in the implementation of our program was reducing the knowledge gap between efficacy and effectiveness. In addition, the established contracts have been characterized for allowing ample margin to the elements of mutual trust, in which the relationship between buyer and supplier was fluid, and the elaboration of the agreements was carried out without excessive problems, contributing to their clear aim to learn.
In short, RSAs are instruments that -duly designed, adapted to local reality and evaluated, under the sincere consensus of both parties- can contribute to aligning the interests of funders and the pharmaceutical industry. The result sought is double. On the one hand, contributing to strengthen pharmacovigilance, thus improving the knowledge of the effectiveness and control of the clinical safety of OD. On the other, favoring the access of these drugs without compromising the sustainability of the system. However, RSAs are not a substitute for traditional agreements, as they may not be appropriate for all medicines nor for all health institutions, due to the previously mentioned limitations -such as insufficient data infrastructure and administrative or initiation burdens23.

