










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroducción
Helicobacter pylori se encuentra en la mitad de la población mundial. Su prevalencia muestra una alta variabilidad según la región geográfica, etnia, raza, edad y factores socioeconómicos, y es alta en países en desarrollo(1). La diversidad de los genotipos de H. pylori parece reflejar la historia de sus hospederos, y se correlaciona con la historia de la migración humana(2). Las cepas de H. pylori de diferentes áreas geográficas muestran características filogeográficas muy específicas, estas pueden ser empleadas para el desarrollo de estudios más complejos(3).
El gen glmM antes conocido como ureC, codifica para una fosfoglucosamina mutasa; y está considerado como “house keeping” y participa directamente en la biosíntesis de la pared celular(4), se ha utilizado para identificar a H. pylori en biopsias gástricas(5). H. pylori exhibe una notable diversidad alélica y variabilidad genética, ya que se recombinan libremente como poblaciones panmícticas, la cual típicamente involucra modificaciones intra-genómica (mutaciones puntuales, recombinaciones, deleciónes y dispariamiento de bases), así como la recombinación inter-genómica, lo que resulta en que los pacientes desarrollen clonas distintas, sin embargo, las diferencias entre estas clonas son mínimas(6). La diversidad de los genotipos de H. pylori parece reflejar la historia de sus hospederos, y se correlaciona con la historia de la migración humana(2).
Las cepas de H. pylori de diferentes áreas geográficas muestran características filogeográficas muy específicas. En los últimos años se han desarrollo nuevas métodos y programas que mediante técnicas moleculares como PCR y análisis genético como DnaSP y PopArt, entre otras, tiene por finalidad conocer la estructura genética de las cepas, estas, han sido un importante avance en el estudio de las enfermedades infecciosas, utilizados para la identificación de agentes patógenos, análisis filogenéticos y genealógicos, para conocer la variabilidad genética aplicados en enfermedades y patologías en diferentes partes del mundo. Además, el análisis genealógico ofrece una visión jerárquica de la evolución de especies, para conocer la relación de parentesco que existe entre los seres vivos. El presente estudio se hizo para conocer la filogeografía con respecto a la variabilidad genética del gen glmM de H. pylori de pacientes con patología gástrica al sur de México.
Material y métodos
Se hicieron crecer 15 cepas de H. pylori en agar Columbia del biobanco del Laboratorio de Investigación Clínica de la Facultad de Ciencias Químico Biológicas, UAGro., todas provenían de pacientes con diagnóstico de enfermedad gástrica (Cuadro 1).
Cuadro 1. Datos de las cepas de H. pylori aisladas de pacientes con patología gástrica incluidos en este estudio
| Cepa | Patolog í a | Genero /edad (años) |
|---|---|---|
| 4191 | Gastritis crónica | Masculino/57 |
| 4192 | Gastritis crónica moderada | Masculino/ 57 |
| 4193 | Gastritis folicular aguda | Femenino/27 |
| 4194 | Gastritis aguda erosiva | Femenino/44 |
| 4195 | Gastritis antral folicular | Masculino/25 |
| 4196 | Gastritis folicular crónica moderada | Masculino/38 |
| 4197 | Gastritis crónica | Femenino/39 |
| 4198 | Gastritis difusa | Femenino/35 |
| 4199 | Gastritis erosiva | Femenino/42 |
| 4200 | Gastritis erosiva | Femenino/42 |
Extracción de DNA cromosomal:
Se obtuvo el DNA de las cepas de H. pylori mediante el método de fenol-cloroformo alcohol isoamílico y se almacenaron a -20°C.
Amplificación por PCR y secuenciación del gene glmM de cepas de H. pylori:
Se utilizó la técnica de PCR para la amplificación del gen glmM empleando los oligonucleótidos: glmMF (5´-CGCGAGCCACAACCCTTTTGAAG-3´), y glmMR (5´-GCTTATCCCCATGCACGATATTC-3‘), con un volumen final de 50µl de la mezcla de reacción (1X buffer PCR (50 mM KCl, 10 mM Tris-HCl [pH 8.3], 1.5% [v/v] Triton X-100), 1.5 mM MgCl2, 200 μM de concentración de cada dNTPs, 50 pmol de primer (glmMF and glmMR), and 1 U de Taq DNA polimerasa)(7). Se utilizó un termociclador MJ Research (Watertonwn, MA, USA), con el siguiente patrón de amplificación: Una temperatura de inicio a 94°C por 5 minutos, por 30 ciclos 94°C por 1 minuto, 54°C por 1:30 minutos, 72°C por 1:30 minutos y extensión final de 10 minutos a 72°C. Los productos de PCR se sometieron a electroforesis en gel de agarosa al 1%, que fueron teñidos con bromuro de etidio y se visualizaron con luz (UV) observándose un producto de aproximadamente 796 pb. Los productos de PCR se purificaron directamente de los geles de agarosa usando el kit de extracción QIAEXII DNA (QIAGEN, Hilden, Germany) y fueron enviados al servicio de secuenciación del Instituto de Biotecnología de la UNAM. Se uso como control positivo DNA genómico de H. pylori ATCC 700392, y control negativo agua.
Determinación de la variabilidad genética y filogeografía:
Se analizaron las secuencias de los productos de PCR de glmM con el programa Chromas y Bioedit para la puntualización y edición de las secuencias, posteriormente se realizó un BLAST en el GenBank para la obtención de 80 secuencias depositadas de glmM, con la finalidad de realizar un multialineamiento con el programa ClustalW2, con la base de datos obtenida de nuestras secuencias y la del GenBank se procesaron en el programa PopArt para la realización de la red de haplotipos, finalmente con el programa DNAsp, se hizo un análisis para determinar la cantidad polimorfismos presentes en el gen glmM(8,9).
Resultados
Caracterización de la población de estudio.
Se analizaron 10 cepas de H. pylori aisladas de pacientes originarios del estado de Guerrero con patologías gástricas, las cuales fueron proporcionadas por el Laboratorio de Investigación en Bacteriología. Los pacientes procedieron de 2 municipios del estado de Guerrero (Chilpancingo de los Bravo, Jaleaca de Catalan), participaron hombres en un 30% y mujeres en 70% con una edad promedio de 42 años.
Análisis filogeográfico: Se analizaron 90 secuencias del gen glmM (10 aislados de pacientes al sur de México y 80 depositadas en el GenBank, con porcentajes de similitud de 96 y 99%). En primera instancia se construyó una red de haplotipos (Figura 1).
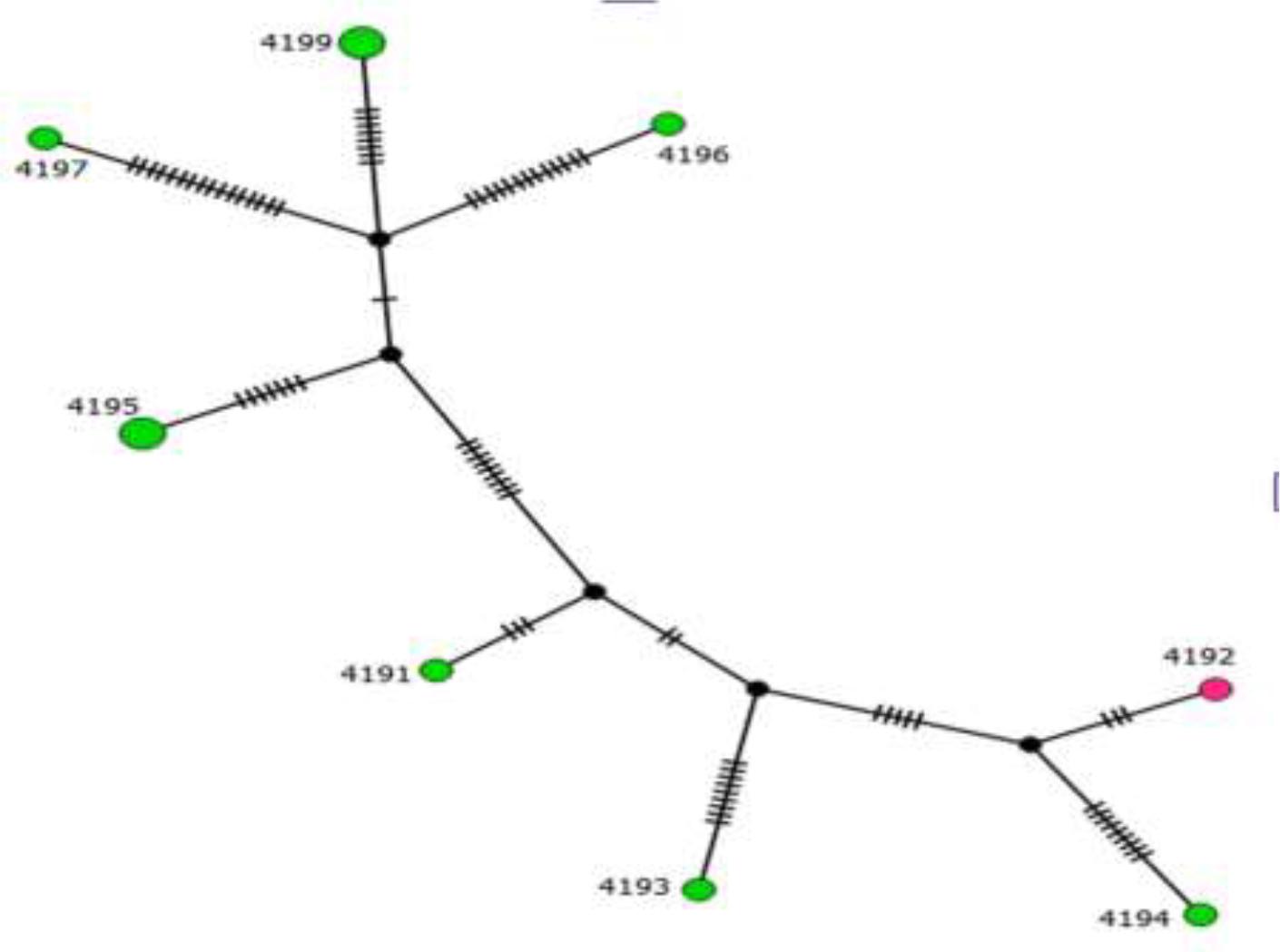
Figura 1. Red de Haplotipos del gen glmM de las 10 secuencias de H. pylori obtenidas de patologías gástricas., en verde se muestran las cepas aisladas de Chilpancingo y en rosa la cepa de Jaleaca, todas del estado de Guerrero, México.
Con el programa PopArt con base a las 10 secuencias obtenidas en el trabajo, en la cual se observan los diferentes haplotipos y la cantidad de pasos mutacionales entre ellos. Posteriormente se realizó otra red de haplotipos comparando las secuencias de Guerrero con las del GenBank (Figura 2).
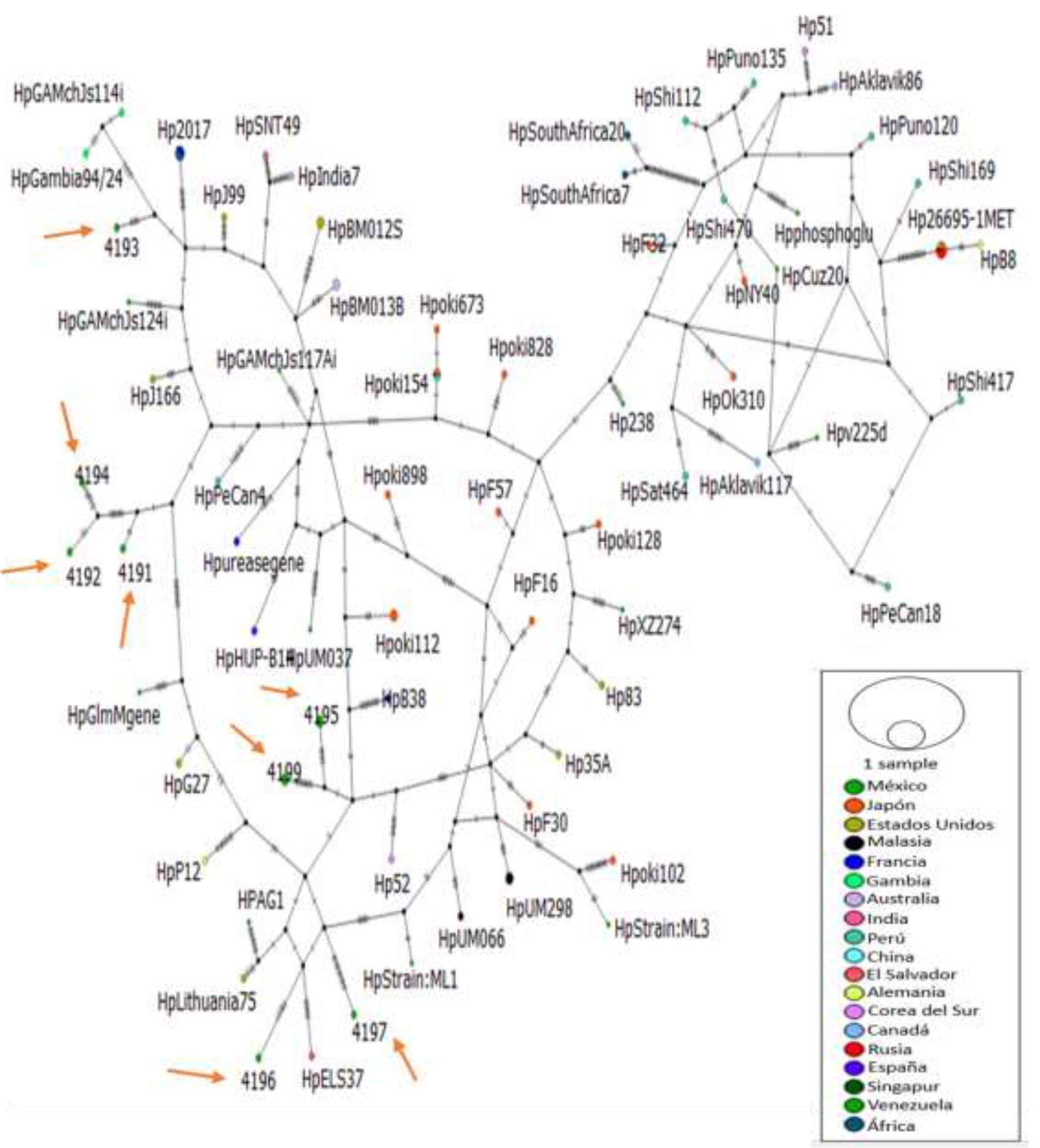
Figura 2. Red de haplotipos del gen glmM de H. pylori. En el grafico anexo a esta red se observa el color asociado a cada país para su identificación en los nodos, las flechas rojas indican la posición de las secuencias de este estudio.
Con las flechas rojas se señalan nuestras secuencias, esta red permite observar las clonas agrupadas en loops con las secuencias del mundo y su gran variabilidad. Con respecto a los parámetros para analizar la diversidad genética se calculó el valor de π (pi) y Θ (theta) los resultados se indican en el Cuadro 2.
Cuadro 2. Comparación de los datos polimórficos, valores de Pi y de theta entre las secuencias de México, con las del mundo.
| México | Mundial | |
|---|---|---|
| Número de secuencias | 10 | 80 |
| Número de sitios | 628 | 628 |
| Número de polimorfismos | 60 | 143 |
| Número de mutaciones | 61 | 150 |
| Número de haplotipos | 9 | 65 |
| Diversidad nucleotídica (P¡) | 0.03416 | 0.03767 |
| Theta (por sitio) de Theta | 0.03316 | 0.04822 |
Discusión
H. pylori representa hoy en día uno de los patógenos más exitosos por el diverso número de estrategias que este microorganismo posee para colonizar el estómago humano, modular la expresión de factores de patogenicidad que orquestan los procesos de daño tisular, sin embargo una de las características más importantes de este microorganismo es una región especifica de su genoma, la zona de plasticidad, la cual es una región hipervariable donde constantemente ocurren mutaciones en los genes codificados en esa región confiriendo un pool de genes que puede variar según las condiciones del hospedero, estos genes auxiliares codifican para diversas propiedades biológicas, la constante variabilidad de estos genes se denomina plasticidad genómica. La plasticidad genómica de H. pylori ha sido estudiada previamente por diversos autores a nivel mundial(6,3), sin embargo, en México y particularmente en Guerrero los estudios realizados son mínimos.
Si bien el estudio de los genes auxiliares es muy importante para tratar de analizar cuáles son posibilidades de desarrollar un cuadro clínico u otro, el estudio de los genes estructurales representa un tipo de estudio arriesgado ya que algunos investigadores consideran que estos genes no sufren grandes cambios al encontrarse en regiones altamente conservadas del genoma por los cual todos los hallazgos de este tipo de investigaciones son relevantes.
En el presente estudio analizo el gen glmM a través de la amplificación de este por PCR, con las secuencias obtenidas de la amplificación del gen de las cepas de H. pylori del estado de Guerrero, fue posible realizar un multialineamiento con una base de datos de 90 secuencias de glmM de H. pylori de diversas parte del mundo, con la finalidad de realizar una red de haplotipos, la cual esquematiza las diferencias entre haplotipos de una especie y las distancias entre estos con pasos mutacionales. Con los datos obtenidos se demuestra que en Guerrero existe una gran diversidad de haplotipos en la especie de H. pylori, encontrando de 10 muestras, 8 haplotipos posiblemente nuevos, estos presentan una gran cantidad de pasos mutacionales entre cada haplotipo, también se observa un determinado grado de homología entre haplotipos de Chilpancingo de los Bravo. Esto puede deberse a que la diversidad genética entre cepas de H. pylori puede originarse por transferencia horizontal(10,11), sin embargo, se puede pensar que los cambios constantes en su genoma pueden ser derivado por una alta recombinación del gen glmM.
Existen diversas condiciones en el microambiente estomacal que pueden afectar el contenido genómico de H. pylori como el pH del ácido gástrico, cantidad de moco gástrico, entre otros(12). Por tanto, los mecanismos que utiliza H. pylori para adquirir o perder genes durante la colonización para adaptarse al ambiente gástrico son diversos(11,13). De este modo podemos decir que la alta variabilidad genética entre cepas de H. pylori se puede deber a la microdiversidad a nivel gen, así como a la macrodiversidad a nivel genoma y claro a la migración poblacional que ha tenido esta bacteria, esto debido a que se observó que los 8 haplotipos encontrados, en una red de haplotipos hecha junto con las 80 secuencias pertenecientes a varios países del mundo, tienen corta distancia relativa en pasos mutacionales, además de formar loops con estos otros haplotipos, permitiendo observar con mayor precisión la distancia que hay entre los haplotipos de Guerrero, lo que nos hace pensar que los haplotipos de México en específico del estado de Guerrero son haplotipos nuevos, que tienen un ancestro con haplotipos de otros continentes y que particularmente estos ocho tienen una mayor plasticidad genómica en comparación con el resto del mundo presentando 60 mutaciones de 148 que tuvieron las 80 secuencias de la base de datos del BLAST.

