










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNefrología (Madrid)
versión On-line ISSN 1989-2284versión impresa ISSN 0211-6995
Nefrología (Madr.) vol.29 no.5 Cantabria 2009
Acidosis tubular renal distal con sordera neurosensorial. Evolución clínica tras 30 años de seguimiento
Distal renal tubular acidosis with neurosensory deafness. Clinical progress after 30 years´ of follow up
Dirección para correspondencia
Sr. Director:
La acidosis tubular renal distal primaria (ATD) es una tubulopatía caracterizada por acidosis metabólica con una orina inapropiadamente alcalina, hipopotasemia e hipercalciuria. Ésta puede ser esporádica o hereditaria, con un patrón autosómico dominante o recesivo. El espectro clínico puede variar mucho en gravedad, desde acidosis leve compensada asintomática con algún cálculo incidental, hasta acidosis grave con retraso en el crecimiento y nefrocalcinosis precoz causante de insuficiencia renal. En general, los pacientes con ATD de herencia dominante tienen un fenotipo más leve que aquéllos con herencia recesiva1.
Existe un subgrupo de pacientes con ATD con herencia recesiva que padece sordera neurosensorial progresiva, causada por mutaciones en el gen que codifica la subunidad B1 de la H+-ATPasa (ATP6V1B1)2. La clínica de esta alteración tubular, además de la sordera, es similar a la descrita en otros tipos de ATD.
Presentamos la evolución clínica de un paciente con ATD con sordera neurosensorial e insuficiencia renal crónica secundaria a nefrocalcinosis, tras 30 años de seguimiento.
Varón de 36 años con cuadro clínico de inicio a los pocos días de vida, consistente en vómitos frecuentes, poliuria, polidipsia, retraso psicomotor, acidosis metabólica hiperclorémica, hipopotasemia, orina alcalina y nefrocalcinosis, estableciéndose el diagnóstico de ATD. Entre los antecedentes familiares, destacaban cosanguineidad de los padres, un hermano afecto de ATD fallecido en accidente laboral y un hermano sano.
El paciente es seguido en nuestras consultas desde los seis años de edad, indicándole tratamiento con citrato sódico y suplementos de potasio oral.
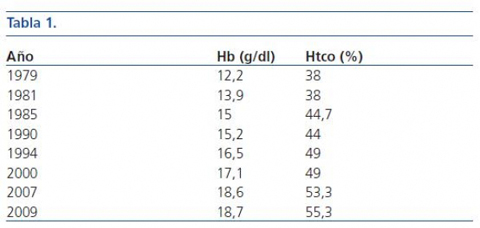
Entre los datos analíticos, destacamos: pH: 7,07-7,33, bicarbonato: 10-26 mmol/L, Cl: 113-124 mEq/L, K: 1,8-3,5 mEq/L. Magnesio, calcio, fósforo, F. alcalina, PTH y Vit D normales. Hb: 12,2-18,7 g/dl, Htco: 38-55,3%. Orina: pH: 7-8, anión Gap: 43 mEq/l, Calcio: 2,5-4,5 mg/kg/día, Fósforo: 6-12 mg/kg/día. Diferencia PCO2 orinaplasma: 2 mmHg (a pH orina: 7,25 y pH plasma: 7,33), índice Cao/Cro: 0,16-0,24.
El paciente realizaba de forma irregular su tratamiento, con abandono frecuente de la medicación y falta de asistencia a sus revisiones. Esto motivó frecuentes ingresos hospitalarios por parálisis muscular hipopotasémica y acidosis metabólica grave con rápida corrección una vez se instauraba tratamiento con bicarbonato y potasio.
A partir de los 16 años de edad, inicia un deterioro lento pero progresivo de la función renal, con un empeoramiento de la nefrocalcinosis (figura 1). A los 19 años de edad, se detecta pérdida de audición y se le diagnostica sordera neurosensorial bilateral, precisando audífono.
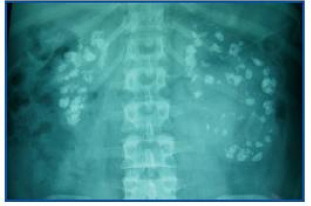
Figura 1
A partir de los 25 años de edad, desarrolla litiasis renal bilateral con complicaciones e ingresos hospitalarios frecuentes en los últimos 10 años. Destacan cólicos renales bilaterales, con desarrollo de calle litiásica espontánea, que en algunas ocasiones se han resuelto con expulsión espontánea de las litiasis y, en otras, han requerido canalización uretral con doble J y litotricia. Varios de ellos han ocasionado obstrucción de la vía urinaria con hidronefrosis y fallo renal agudo sobreañadido a su enfermedad renal crónica, con resolución tras la expulsión litiásica.
En las últimas revisiones se constata un aumento progresivo de la nefrocalcinosis, y de forma paralela existe un aumento progresivo de Hb y Htco con poliglobulia actual (tabla 1) y nivel de EPO normal (11,6 mU/ml).
Actualmente, el paciente mantiene un aclaramiento de creatinina estabilizado en torno a 45 ml/min.
Para nosotros, la importancia de este caso radica en mostrar las principales complicaciones que pueden derivar de esta enfermedad y que se han presentado en nuestro paciente: 1) parálisis muscular hipopotasémica, consecuencia del abandono frecuente del tratamiento; 2) litiasis renal bilateral, que a su vez ha producido fallo renal agudo obstructivo, requiriendo manipulaciones múltiples de la vía urinaria e, incluso, ha llegado a provocar calle litiásica espontánea, algo poco descrito en este tipo de pacientes3; 3) poliglobulia secundaria probablemente a la nefrocalcinosis, ya que la hipoxia tisular induciría un aumento en la producción de EPO que, aunque en nuestro caso está en rango normal, puede ser inapropiadamente alta para una hemoglobina tan elevada. Esta complicación muy poco comunicada en la literatura como asociada a la ATD4,5; 4) insuficiencia renal crónica, que en nuestro caso, y a pesar de las múltiples complicaciones, progresa de forma muy lenta (20 años desde que inicia el descenso en el aclaramiento de creatinina) y que se mantiene estable en los últimos años.
Concluimos que en la ATD es muy importante supervisar el cumplimiento del tratamiento para evitar complicaciones, algunas potencialmente graves y perfectamente evitables, así como prestar atención al desarrollo de poliglobulia secundaria a la nefrocalcinosis, la cual podría favorecer el desarrollo de eventos trombóticos en estos pacientes. A pesar de todo, la progresión de la insuficiencia renal sigue una evolución muy lenta en estos 30 años de seguimiento.
R. López Hidalgo, A. Polo Moyano, M. Manjón Rodríguez, S. Cerezo Morales
Servicio de Nefrología. Hospital Universitario San Cecilio. Granada
Bibliografía
1. Karet FE. Inherited distal renal tubular acidosis. J Am Soc Nephrol 2002;13:2178-84. [ Links ]
2. Karet FE, Finberg KE, Nelson RD et al. Mutations in the gene encoding B1 subunit of H -ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 1999;21(1):84-90. [ Links ]
3. Peces R. Steinstrasse due to distal renal tubular acidosis with sensorineural deafness. Nephrol Dial Transplant 2000;15:1251-2. [ Links ]
4. Agroyannis B, Koutsikos D, Tzanatos- Exarchou H, et al. Erythrocytosis in type I renal tubular acidosis with nephrocalcinosis. Nephrol Dial Transplant 1992;7:365-9. [ Links ]
5. Matsukura H, Satoh H, Arai M, et al. Secondary erythrocytosis associated with distal renal tubular acidosis. Clinical Nephrology 2004;62(5):397-9. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Raquel López Hidalgo
Servicio de Nefrología.
Hospital Universitario San Cecilio. Granada
rlopezh@senefro.org

