










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNefrología (Madrid)
versión On-line ISSN 1989-2284versión impresa ISSN 0211-6995
Nefrología (Madr.) vol.34 no.3 Cantabria 2014
https://dx.doi.org/10.3265/Nefrologia.pre2014.Jan.12335
REVISIONES
Hiperoxaluria primaria
Primary hyperoxaluria
Víctor Lorenzo1, Armando Torres1, Eduardo Salido2
1Servicio de Nefrología. Hospital Universitario de Canarias. San Cristóbal de La Laguna, Santa Cruz de Tenerife
2Servicio de Anatomía Patológica. Facultad de Medicina. San Cristóbal de La Laguna, Santa Cruz de Tenerife
Este trabajo ha recibido financiación de: PROYECTO FIS PI070963 (Instituto de Salud Carlos III, y Fondos FEDER), RedInRen RD12/0021/0008, y SAF2011-23933 (proyecto del MINECO).
Dirección para correspondencia
RESUMEN
La hiperoxaluria primaria (HOP) se debe a un desorden metabólico hereditario autosómico recesivo, del metabolismo del glioxalato, que causa una producción excesiva de oxalato. El trastorno más frecuente y grave se debe al déficit enzimático de alanin:glioxalato aminotransferasa (HOP tipo I) específico en el peroxisoma hepático. Dado que el oxalato no se metaboliza en los humanos y se elimina por vía renal, el riñón es el primer órgano afectado, dando lugar a la aparición de litiasis de repetición, nefrocalcinosis e insuficiencia renal precoz. Con la progresión de la insuficiencia renal, especialmente en pacientes sometidos a hemodiálisis (HD), el oxalato cálcico se deposita masivamente en los tejidos, denominándose a esto último oxalosis. El diagnóstico se basa en los antecedentes familiares, la presencia de urolitiasis y/o nefrocalcinosis, hiperoxaluria, depósitos tisulares de oxalato formando granulomas en formas avanzadas, análisis molecular de ADN y análisis enzimático si procede. Se requiere una alta sospecha diagnóstica, por lo que, desafortunadamente, en muchos casos se diagnostica tras su recidiva en el trasplante renal. El manejo conservador de la enfermedad (alta ingesta líquida, piridoxina e inhibidores de la cristalización) debe ser precoz, para retrasar el daño renal. El tratamiento con diálisis es inefectivo para depurar el exceso de oxalatos. Tras el trasplante renal suele observarse una rápida aparición de los depósitos de oxalato en el injerto y los resultados de esta técnica, salvo excepciones, son desalentadores. El trasplante hepático anticipado, o simultáneo con el trasplante renal cuando ya existe daño irreversible de este órgano, es la opción terapéutica de elección para corregir la enfermedad de base y suprimir la sobreproducción de oxalatos. Dada la condición de enfermedad rara y su heterogeneidad genética y clínica, no es posible obtener evidencias a través de ensayos clínicos aleatorizados. Por lo tanto, las recomendaciones las establecen grupos de expertos apoyados en publicaciones de acreditado rigor científico. En este sentido, un grupo de expertos europeos (OxalEurope) ha elaborado unas recomendaciones diagnósticas y terapéuticas publicadas en 2012.
Palabras clave: Hiperoxaluria primaria, Estados hiperoxalúricos, Oxalosis, Litiasis renal, Trasplante hepato-renal.
ABSTRACT
Primary hyperoxaluria (PH) occurs due to an autosomal recessive hereditary disorder of the metabolism of glyoxylate, which causes excessive oxalate production. The most frequent and serious disorder is due to enzyme deficit of alanine-glyoxylate aminotransferase (PH type I) specific to hepatic peroxisome. As oxalate is not metabolised in humans and is excreted through the kidneys, the kidney is the first organ affected, causing recurrent lithiasis, nephrocalcinosis and early renal failure. With advance of renal failure, particularly in patients on haemodialysis (HD), calcium oxalate is massively deposited in tissues, which is known as oxalosis. Diagnosis is based on family history, the presence of urolithiasis and/or nephrocalcinosis, hyperoxaluria, oxalate deposits in tissue forming granulomas, molecular analysis of DNA and enzyme analysis if applicable. High diagnostic suspicion is required; therefore, unfortunately, in many cases it is diagnosed after its recurrence following kidney transplantation. Conservative management of this disease (high liquid intake, pyridoxine and crystallisation inhibitors) needs to be adopted early in order to delay kidney damage. Treatment by dialysis is ineffective in treating excess oxalate. After the kidney transplant, we normally observe a rapid appearance of oxalate deposits in the graft and the results of this technique are discouraging, with very few exceptions. Pre-emptive liver transplantation, or simultaneous liver and kidney transplants when there is already irreversible damage to the kidney, is the treatment of choice to treat the underlying disease and suppress oxalate overproduction. Given its condition as a rare disease and its genetic and clinical heterogeneity, it is not possible to gain evidence through randomised clinical trials. As a result, the recommendations are established by groups of experts based on publications of renowned scientific rigour. In this regard, a group of European experts (OxalEurope) has drawn up recommendations for diagnosis and treatment, which were published in 2012.
Key Words: Primary hyperoxaluria, Hyperoxaluric states, Oxalosis, Renal lithiasis, Liver-renal transplantation.
Introducción
La hiperoxaluria primaria (HOP) es un desorden metabólico hereditario autosómico recesivo del metabolismo del glioxalato, que cursa con una producción excesiva de oxalato. El trastorno más frecuente se debe al déficit enzimático de alanin:glioxalato aminotransferasa (HOP tipo I) específico en el peroxisoma hepático1-4. La incidencia de HOP es difícil de estimar, dado que muchos casos son reconocidos tardíamente o bien nunca son identificados. Tiene una prevalencia estimada de 1-3 por millón de población y una tasa de incidencia de aproximadamente 1:100000 nacidos vivos5,6. Se han descrito tasas mayores en poblaciones históricamente aisladas, como en las Islas Canarias, por un efecto fundador7. Afecta a menos del 1% de la población pediátrica con enfermedad renal terminal, siendo más frecuente en poblaciones donde la consanguinidad es mayor8.
Metabolismo del ácido oxálico
El oxalato es un ácido dicarboxílico (C2O4H2, peso molecular 90Da) que proviene principalmente del metabolismo endógeno y solo una pequeña parte de la dieta. Se produce en el hígado a partir del glioxalato. Esta es una molécula generada en el metabolismo intermedio de la glicina, hidroxiprolina y glicolato. La detoxificación del glioxalato se realiza mayormente por la alanin-glioxalato amino transferasa (AGT) en el peroxisoma del hepatocito humano, convirtiendo el glioxalato en glicina. La vitamina B6 actúa como cofactor. En condiciones normales, solo parte del glioxalato es transformado en oxalato por la lactato dehidrogenasa (LDH)9 (figura 1). El oxalato no puede ser metabolizado por los mamíferos, no se liga a las proteínas y no se metaboliza. Es filtrado por el glomérulo y también secretado por el túbulo, eliminándose sin cambios por vía renal. La eliminación urinaria es normalmente inferior a 45mg/día o 0,45mmoles/l/1,73m2 por día (mg se convierten a mmoles multiplicando por 0,01136)4,10,11.

Figura 1. Metabolismo del ácido oxálico.
AGT: alanin-glioxalato amino transferasa; LDH: lactato dehidrogenasa; Vit: vitamina.
Enfermedad por exceso de producción de oxalato.
Los defectos genéticos de las enzimas que metabolizan el glioxalato dan lugar a la sobreproducción hepática de oxalato, es decir, que estamos ante una enfermedad por exceso de producción. Se denomina HOP y el término de oxalosis se aplica a los depósitos tisulares.
Clasificación de los estados hiperoxalúricos
La causa principal y más seria de hiperoxaluria son los defectos enzimáticos hereditarios, denominados HOP. En estos casos la excreción urinaria de oxalato es >45mg/día/1,73m2 y con frecuencia >801,12. Además, existen otras situaciones denominadas hiperoxalurias secundarias (HOS)13. Las causas de HOS son la ingesta abusiva de precursores del oxalato y el aumento de la absorción intestinal (hiperoxaluria entérica). En general, son formas menos graves (<70mg/dl) y no suelen desarrollar oxalosis sistémica. En la tabla 1 se describe la clasificación de estados hiperoxalúricos.
Tabla 1. Clasificación de los estados hiperoxalúricos. 
Hiperoxaluria primaria
Se trata de un desorden metabólico hereditario autosómico recesivo del metabolismo del glioxalato que resulta de una elevada producción hepática de oxalato2,14,15. Se han descrito tres tipos de trastornos moleculares. Los genes implicados son la alanin-glioxalato aminotransferasa (AGXT) para la HOP tipo 1 (HOP-I), que representa el 80% de los pacientes que sufren HOP; la glioxalato reductasa/hidroxipiruvato reductasa (GRHPR) localizado en el cromosoma 10, para la HOP-II; y la 4-OH-2-oxoglutarato aldosala (HOGA1, también conocido como DHDPSL) localizado en el cromosoma 9, para la HOP-III16.
Hiperoxaluria primaria tipo I
La HOP-I es causada por el déficit de la AGT, enzima que cataliza la transaminación de L-alanina y glioxalato a piruvato y glicina. Se expresa específicamente en el hígado de los humanos y se localiza en el peroxisoma4,17-20. Su déficit produce incremento de glicolato y oxalato21. La clonación del AGXT cDNA y el conocimiento de su estructura tridimensional ha proporcionado información relevante sobre las funciones de la proteína y el efecto de los cambios en la secuencia de aminoácidos en la mayoría de las más de 150 mutaciones descritas de la HOP22-24.
Estas mutaciones dan lugar a distintas expresiones fenotípicas de la enfermedad por otros tantos mecanismos moleculares4. Se han descrito, básicamente, cuatro mecanismos distintos:
1. Mistargeting mitocondrial: podemos traducir este término como «tráfico proteico intracelular erróneo», sin precedentes en otra enfermedad genética humana. La proteína se localiza en la mitocondria, donde es inactiva, en lugar del peroxisoma25-27. Una de las formas más frecuentes es la Gly170Arg27, descrita como missense mutation (cambio de un nucleótido simple en un codón que codifica un aminoácido diferente). Otra mutación missense, F152I, también produce este tráfico intracelular erróneo y el déficit de AGT peroxisomal.
2. Agregación proteica: es un resultado frecuente dentro de las missense mutation en las enfermedades conformacionales. La más común es la mutación G41R28 y la Ile244Thr20. Esta última variante se asocia a agregación y acelerada degradación de la AGT por interacción con «chaperones» moleculares (brevemente, son proteínas que contribuyen al plegamiento y conformación tridimensional de otras proteínas). En esta mutación, para que la expresión fenotípica tenga lugar, se requiere la presencia además de un polimorfismo del Pro11Leu en el mismo alelo. Esta es la alteración descrita en la comunidad canaria23, siendo la mayoría de las familias afectas procedentes de la isla de La Gomera.
3. Abolición de la actividad catalítica22: mecanismo común a muchos errores del metabolismo que afectan a genes que codifican enzimas. Varias missense mutation (G82E, G41R, F152I) alteran la actividad catalítica de la AGT.
4. Defecto de síntesis29: la más común en este grupo es c.33Cdup, definiéndose como nonsense mutation, es decir, mutación que resulta en la aparición de un codón de parada, dando lugar a una proteína truncada, habitualmente no funcionante.
La investigación de los mecanismos moleculares, sin duda, derivará en el mejor conocimiento de las correlaciones genotipo-fenotípicas y en la investigación de nuevas opciones terapéuticas.
Hiperoxaluria primaria tipo II
La HOP-II o L-glycerico aciduria representa aproximadamente el 10% de las HOP. El déficit de la enzima glioxalato/hidroxihipurato reductasa (GRHPR) induce un aumento de excreción urinaria de oxalato y L-glicerato. Estos pacientes sufren una elevación urinaria de oxalato más leve que en la HOP-I y son pocos los que desarrollan enfermedad renal terminal30.
Hiperoxaluria primaria tipo III
Ocurre en el 10% de los casos de HOP; la mutación del gen HOGA1 produce el déficit de la enzima mitocondrial 4-hidroxi-2-oxoglutarato aldolasa que desdobla el 4-hidroxi-2-oxoglutarato en piruvato y glioxalato, que a su vez es transformado en oxalato por la LDH. Se presenta con un amplio rango de eliminación urinaria de oxalato, pero la progresión a enfermedad renal terminal no ha sido descrita16.
Hiperoxaluria secundaria
En la tabla 1 se resumen las causas de HOS.
Hiperoxaluria entérica
El oxalato proveniente de la dieta puede absorberse a lo largo de todo el tubo digestivo, tanto por difusión pasiva como por transporte activo. Todas las situaciones que producen malabsorción de sales biliares y ácidos grasos en el íleon pueden aumentar la absorción de oxalato31. Esto es debido a que al llegar al colon aumentan la permeabilidad de la mucosa colónica al oxalato, y además el calcio se une a los ácidos grasos, dejando oxalato libre y soluble, fácilmente absorbible32-36. En estudios experimentales también se ha demostrado que la acidificación del colon con lactulosa aumenta significativamente la excreción urinaria de oxalatos37.
De esta forma, las enfermedades inflamatorias intestinales que cursan con esteatorrea31,35 y las resecciones ileales amplias38-41 pueden desarrollar hiperoxaluria y litiasis, siempre que el colon esté intacto35. El aumento de prevalencia de litiasis renal en casos de fibrosis quística de páncreas, además de la hipocitraturia, se ha relacionado con el incremento de excreción renal de oxalato. De forma anecdótica, se han descrito casos de pérdida del injerto renal por oxalosis en pacientes trasplantados que desarrollaron esteatorrea secundaria a trastornos biliopancreáticos43.
Más recientemente, la hiperoxaluria y la nefrolitiasis han sido una complicación descrita en pacientes sometidos a cirugía bariátrica para el tratamiento de la obesidad mórbida44-46, requiriendo incluso diálisis crónica en dos casos46. Por tanto, es recomendable la vigilancia periódica de la oxaluria en el posoperatorio precoz y a largo plazo de estos pacientes.
Otro factor importante que puede influir en la absorción de oxalato es la descolonización colónica con bacterias metabolizadoras de oxalato, siendo el Oxalobacter formigenes el mejor estudiado (ver más adelante)47. La ausencia de estas bacterias produce hiperoxaluria por aumentar la absorción de oxalato. El empleo de antibióticos se ha postulado como un factor de su erradicación.
Ingesta abusiva de precursores
La ingestión intencional o accidental de etilenglicol, cuyo uso más frecuente es como anticongelante de motores, puede desarrollar crisis hiperoxalúricas graves48. La toxicidad del etilenglicol está relacionada con su biotransformación a ácido glicólico, causando acidosis normoclorémica grave, hiperoxaluria y oxalosis49,50.
El ácido ascórbico (vitamina C) es un precursor metabólico del oxalato51, habiéndose demostrado que su empleo abusivo puede inducir sobresaturación de oxalato cálcico y las complicaciones derivadas de su precipitación52-54.
Presentación clínica
Existe una considerable heterogeneidad en el patrón de presentación de la enfermedad y de su progresión a la insuficiencia renal55. Esta gran variabilidad clínica no se relaciona bien con las mutaciones del gen o con el grado de actividad enzimática residual. Además, hay factores inhibidores y promotores de la cristalización del oxalato que modulan el patrón genético favoreciendo su gran heterogeneidad clínica.
Las formas más graves aparecen en la HOP-I, habiéndose descrito formas infantiles que debutan en los primeros meses de vida y presentan una elevada mortalidad precoz. Las más comunes aparecen en torno a la segunda década de la vida y muchos pacientes se tratan como una litiasis recidivante, pasando desapercibido el diagnóstico durante años. De hecho, con frecuencia (30-60%, según las series) el diagnóstico de HOP se realiza tras la recidiva de depósitos de oxalatos en el trasplante renal3,7,56,57. Esto es más desafortunado aún cuando se trata de un donante vivo, como ocurrió en un caso de nuestra serie de enfermos7. Existen variantes menos agresivas que se diagnostican en la edad adulta por la presencia de litiasis y/o nefrocalcinosis, que cursan con una supervivencia prolongada, aun en diálisis. Incluso hay pacientes de edad avanzada que no llegan a desarrollar enfermedad renal terminal2,3.
En los casos de HOP-II la enfermedad suele ser menos agresiva58 y solo una pequeña proporción de pacientes (aproximadamente el 20%) desarrolla enfermedad renal terminal. Los casos de HOP-III son aún menos frecuentes (10%) y más benignos, aunque la experiencia es más limitada en ellos16,59.
Las manifestaciones clínicas destacadas son nefrolitiasis recidivante, nefrocalcinosis, hematuria, infecciones urinarias e insuficiencia renal de rápida evolución. Conviene destacar que la nefrocalcinosis es característica y que aparece acompañando a la nefrolitiasis o bien de forma aislada (figura 2).
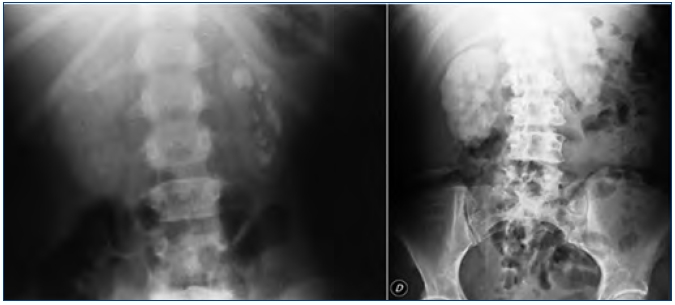
Figura 2. Litiasis renal múltiple y nefrocalcinosis en
pacientes con hiperoxaluria primaria.
Radiografía simple de abdomen.
Aunque estamos ante una enfermedad con señas de identidad bien definidas, dada su rareza, lo habitual es que ante pacientes con nefrolitiasis de repetición el médico responsable investigue causas de HOS y las más comunes de nefrolitiasis. El diagnóstico diferencial debe apoyarse en las siguientes premisas:
- Interrogar y analizar las posibles causas de HOS: ingesta de precursores del glioxalato y enfermedades inflamatorias intestinales o resecciones intestinales amplias, incluida la cirugía bariátrica.
- Litiasis metabólicas: hipercalciuria (>4mg/kg/día sin hipercalcemia, o >0,15mg/dl filtrado glomerular [FG]); hipocitraturia (<300mg/24 horas, o <0,17mg/dl FG) e hiperuricosuria (>750-800mg/24 horas o >0,45mg/dl FG). No cursan con nefrocalcinosis ni desarrollan insuficiencia renal precoz.
- Estados hipercalcémicos: sarcoidosis, hiperparatiroidismo primario, hipervitaminosis D, síndrome leche-alcalinos. Pueden cursar con litiasis y nefrocalcinosis, pero obviamente su rasgo diferencial es la hipercalcemia, que no ocurre en la HOP.
- Acidosis renal tubular tipo 1: pueden desarrollar litiasis y nefrocalcinosis si cursan con hipercalciuria. No obstante, su seña de identidad es la acidosis metabólica hiperclorémica, la hipopotasemia y el desarrollo de insuficiencia renal es raro.
- Cistinuria: cristales hexagonales característicos en el sedimento, menos radiopacos, no desarrollan nefrocalcinosis y la presentación de enfermedad renal avanzada o terminal es rara.
Finalmente, basándonos en los datos clínicos, en la tabla 2 se describen las situaciones clínicas en que debe investigarse el diagnóstico de HOP, siempre en ausencia de datos que sugieran hiperoxaluria entérica o ingesta anómala de precursores.
Tabla 2. Parámetros clínicos ante los que debe
investigarse el diagnóstico de hiperoxaluria primaria. 
HOP: hiperoxaluria primaria.
Daño tisular en la hiperoxaluria primaria: oxalosis
El riñón es el primer órgano afectado con agregados de oxalato cálcico en el espacio urinario (urolitiasis) y en el tejido renal (nefrocalcinosis) (figura 3), donde se desarrolla importante fibrosis intersticial e insuficiencia renal1,60,61. Una vez que el FG cae por debajo de 30-40ml/min/1,73m2, la eliminación urinaria no es capaz de mantener la oxalemia dentro de la normalidad (<6μmol/l) y puede superar el umbral de saturación de oxalato cálcico cuando los niveles son mayores de 30μmol/l, produciéndose depósitos tisulares en forma de monohidrato y dihidrato62-64. A este fenómeno se lo denomina oxalosis y da origen a una importante respuesta inflamatoria en forma de granulomas alrededor de los cristales.

Figura 3. Depósitos de oxalato cálcico en el espacio
urinario y en el tejido renal
(HE, microscopía de polarización, 200X).
Se han identificado depósitos extrarrenales de oxalato cálcico en la mayoría de los tejidos y órganos, tales como retina, miocardio, vasos sanguíneos, piel, hueso y sistema nervioso3,61,62,65, si bien ocurren preferentemente donde la concentración de calcio es mayor. El desarrollo de cardiomiopatía y trastornos de conducción, vasculopatía con frecuentes necrosis distales66, retinopatía, sinovitis o enfermedad ósea de alto remodelado65 es una complicación tardía grave que conduce a una mortalidad precoz.
Una vez en hemodiálisis, los depósitos tisulares de oxalato progresan de forma espectacular, siendo el tejido óseo el órgano más afectado65,67. Los enfermos cursan con dolores óseos progresivos, invalidez y deformidades esqueléticas. En el examen radiológico, destaca el marcado incremento de la densidad ósea, el hueso pierde la trama ósea normal y se observan lesiones de alto remodelado, tipo osteítis fibrosa, pero de una gravedad inusual65,68.
El estudio y seguimiento con biopsia ósea nos permitió conocer el punto de comienzo de la enfermedad ósea y seguir su progresión en hemodiálisis65. En la etapa prediálisis las manifestaciones clínicas o radiológicas de enfermedad ósea están ausentes, aunque pueden aparecer focos incipientes de oxalosis peritrabecular en el estudio histológico (figura 4 A). Este parece ser el sitio inicial de depósito, probablemente por ser la zona de mayor concentración de calcio. Al cabo de uno o dos años las lesiones evolucionan con inusitada gravedad. Existen evidencias de que la reacción celular al depósito de cristales es la que activa y acelera el remodelado óseo, simulando un hiperparatiroidismo. Las células gigantes multinucleadas que engloban los cristales pertenecen a la serie macrófago-monocítica, al igual que los osteoclastos. La acción de este grupo celular sobre las trabéculas óseas adyacentes a los granulomas pondría en marcha la resorción ósea. Por el normal acoplamiento osteoclasto-osteoblasto, estos se activan generando osteoide predominantemente no laminar. Los estudios dinámicos revelan una captación difusa de tetraciclinas en áreas de osteoide no laminar. Los cristales aparecen fundamentalmente en el espacio medular e invadiendo la superficie trabecular. Se agrupan en forma de estrella o roseta y a su alrededor se produce una reacción granulomatosa a cuerpo extraño con células gigantes multinucleadas, macrófagos, fibroblastos y fibrosis medular (figura 4 B). Al examen con luz polarizada, estos cristales son altamente refringentes, apareciendo fragmentados en subunidades (figura 4 C)65. Los niveles séricos de fosfatasa alcalina y parathormona suelen aparecer solo discretamente aumentados, en contraste con los casos del hiperparatiroidismo grave.

Figura 4. Oxalosis ósea.
A) Depósitos peritrabeculares de oxalato cálcico (azul de toluidina, microscopía de polarización, 40X). B) Cristales agrupados
en forma de estrella o roseta, a cuyo alrededor se produce una reacción granulomatosa a cuerpo extraño con células gigantes
multinucleadas, macrófagos, fibroblastos y fibrosis medular (Goldner, 100X). C) Cristales altamente refringentes en el espacio
medular con reacción granulomatosa circundante (azul de toluidina, microscopía de polarización, 60X).
También hemos documentado depósitos de oxalato en otros tejidos. En un paciente con HOP y polineuropatía grave constatamos por microscopía electrónica depósitos de oxalato en las células de Schwann y en las vainas de mielina del nervio periférico a los pocos meses de comenzar a dializarse (figura 5). En otro paciente con patología cardíaca, el diagnóstico lo proporcionó la presencia de cristales de oxalato en biopsia subendocárdica (figura 6), que después fue confirmado con el análisis de ADN. En otros dos pacientes con HOP en hemodiálisis hemos observado una resistencia primaria y absoluta al tratamiento con eritropoyetina, a pesar del empleo de elevadas dosis de esta hormona. La oxalosis ósea grave produce una extensa ocupación de la médula ósea por tejido granulomatoso y fibrosis que afecta la eritropoyesis al desplazar el tejido hematopoyético69,70. Sin embargo, hay órganos como el hígado que no se han visto afectados, probablemente porque su concentración intracelular de calcio sea menor.
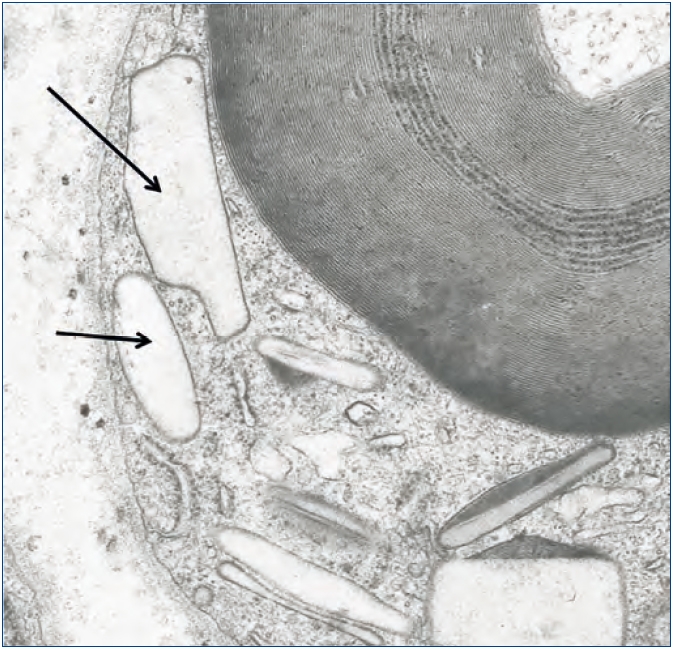
Figura 5. Depósitos de oxalato en células de Schwann.
Células de Schwann con cuerpos traslúcidos de oxalato
(flecha), algunos de ellos asociados a estructuras densas
(lisosomas) (microscopía electrónica X6000).
Con permiso de Lorenzo et al.: «Morfología normal y patológica».

Figura 6. Depósitos de oxalato en tejido subendocárdico
(HE, microscopía de polarización, 20X).
Métodos diagnósticos
El diagnóstico precoz es crucial para prevenir la enfermedad renal terminal. Como apuntábamos previamente, una meticulosa historia familiar, junto a la determinación de oxaluria, es fundamental en casos de sospecha de HOP. La exploración del sedimento puede proporcionar información adicional con la presencia de cristales de oxalato cálcico monohidrato (whewellita, CaCO4.H2O) en forma de «pesa de gimnasio» y que debe diferenciarse de los típicos cristales romboidales de oxalato cálcico dihidrato71. Si la sospecha diagnóstica persiste, la determinación de glicolato en orina de 24 horas y la oxalemia pueden proporcionar información adicional.
Aunque los depósitos tisulares de oxalato suelen ser más tardíos, deben investigarse los efectos sobre el corazón (electrocardiograma y ecocardiograma), el fondo de ojo y el esqueleto55. La tomografía computarizada puede ser de gran ayuda para evaluar la extensión de las calcificaciones y los depósitos tisulares de oxalato.
El estudio histológico que demuestre la presencia de cristales de oxalato, especialmente en la biopsia renal, puede ser de utilidad en casos dudosos.
El análisis genético es obligado para confirmar el diagnóstico y tipificar la variante mutacional. Dado que solamente se requiere ADN de sangre periférica, el estudio de las mutaciones comunes se ha convertido en el siguiente escalón diagnóstico, una vez constatada la hiperoxaluria. La actividad enzimática de la AGT en tejido hepático puede servir de ayuda si el estudio genético no es esclarecedor72. El consejo genético y el diagnóstico prenatal mediante el análisis mutacional de los padres son recomendaciones pertinentes.
Recientemente ha sido publicado por las guías europeas un algoritmo para dirigir el diagnóstico de la HOP73. Por nuestra parte, en la figura 7 se expone un algoritmo diagnóstico orientativo, simplificado, que debe adecuarse a las circunstancias clínicas y demográficas individuales y a las disponibilidades del medio.
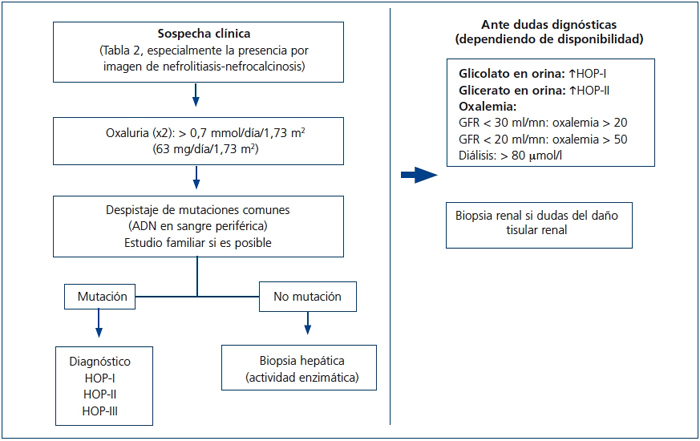
Figura 7. Algoritmo diagnóstico de la hiperoxaluria primaria.
GFR: tasa de filtrado glomerular; HOP: hiperoxaluria primaria.
Métodos bioquímicos
La determinación urinaria de oxalato es el primer paso en el diagnóstico de HOP. Una oxaluria superior a 45mg/día (>0,5mmol/1,73m2/día) en, al menos, dos muestras de orina de 24 horas es característica de HOP, una vez que han sido excluidas causas de HOS1,12. Muestras superiores a 80-90mg/día son muy sugerentes de HOP-I55.
La elevación de glicolato es propia de la HOP-I y, aunque tiene baja especificidad y sensibilidad diagnóstica, valores de glicolato urinario superiores a 45mg/día (>0,5mmol/1,73m2/día) son sugerentes de HOP-I12,55,74.
La elevación de niveles de L-glicerato es indicativa de HOP-II. La extrema rareza de las formas de HOP-II hace que la determinación de glicerato en orina solo pueda ser realizada en centros altamente especializados en el análisis de ácidos orgánicos75.
En lactantes y niños pequeños, donde la recogida de orina es difícil, una muestra aislada por la mañana puede ser orientativa. Estos resultados deben interpretarse conforme a los valores de referencia para la edad del paciente1,75,76.
Los valores séricos de oxalato permanecen en el rango de la normalidad hasta que el filtrado renal no cae por debajo de 45ml/min aproximadamente. Dada la amplia variabilidad de valores, no se dispone de un umbral fiable para establecer el diagnóstico. Ante valores superiores a 50µMol/l debe investigarse la enfermedad y valores superiores a 100μMol/l son muy sugerentes de HOP55,62,76,77.
Diagnóstico histológico
La presencia de abundantes cristales de oxalato en la biopsia renal es indicativa de HOP, especialmente si aparecen en el intersticio, rodeados de una fuerte reacción inflamatoria en forma de granulomas de cuerpo extraño. No es infrecuente que estos depósitos aparezcan en un riñón trasplantado y den el diagnóstico de la enfermedad de base tardíamente7. En casos avanzados de pacientes en diálisis con imágenes de enfermedad ósea de alto remodelado, la biopsia ósea ha proporcionado el diagnóstico definitivo, mostrando las típicas reacciones granulomatosas rodeando los depósitos de oxalato7,65.
Análisis del ADN
El diagnóstico de HOP debe ser confirmado con el análisis del ADN del gen AGT (se conoce con el símbolo AGXT), que informa además sobre el tipo de mutación y polimorfismos que afectan al paciente18,20,23,78-80. Cuando nos enfrentamos a posibles mutaciones no comunes, es necesario el análisis genético de familiares directos. Dado que el análisis del ADN es un método no invasivo, se ha convertido en una exploración diagnóstica de primera línea ante la sospecha clínica y bioquímica de HOP, siendo utilizado además para el test prenatal y el diagnóstico de otros miembros de la familia, una vez conocida la mutación.
Diagnóstico enzimático
La biopsia hepática está indicada cuando no se encuentra la mutación en los genes AGXT, GRHPR o HOGA1 para excluir completamente las variantes conocidas de la HOP. Los niveles de actividad enzimática AGT en pacientes de HOP-I es bastante variable, sobre todo en los afectos de la mutación mistargeting, donde la AGT está presente en la mitocondria. En este caso el análisis inmunohistoquímico puede proporcionar la localización subcelular de la enzima81,82.
Diagnóstico prenatal
Dado que la AGT solo se expresa en el hígado, las determinaciones enzimáticas llevadas a cabo en el líquido amniótico no son útiles. Sin embargo, empleando el análisis de ADN en mujeres embarazadas y familiares se puede conocer el diagnóstico prenatal de las mutaciones más comunes83. Este procedimiento diagnóstico es el que más puede ganar al hacer posible el análisis del gen AGXT a partir de células del líquido amniótico o por biopsia de vellosidades coriales. Es previsible que estas técnicas moleculares se conviertan en el principal método diagnóstico de la HOP, al poder llevarse a cabo con ADN de cualquier célula del organismo y proporcionar información menos ambigua que la determinación de actividad enzimática en la biopsia hepática.
Tratamiento
Medidas generales
El tratamiento inicial debe ser precoz y se dirige a disminuir la saturación urinaria de oxalato cálcico, aumentando la ingesta de líquidos y empleando inhibidores urinarios de la cristalización84. La ingesta de líquidos debe superar los 3 l/m2/día85 y el pH urinario debe mantenerse entre 6,2 y 6,8. Estas medidas generales son aplicables a todos los estados hiperoxalúricos y su eficacia dependerá de la magnitud del problema, por lo que en los casos de HOP severos suele ser muy limitada55.
Las pautas recomendadas de los inhibidores de la cristalización son las siguientes:
- Citrato potásico: forma complejos con el calcio, disminuyendo la precipitación de oxalato cálcico, y aumenta el pH urinario86. Se recomienda una dosis diaria de citrato potásico de 0,1-0,15g/kg de peso12,55,86,87. En casos de insuficiencia renal el citrato potásico debe ser sustituido por citrato sódico88. Las tiazidas (en combinación con el citrato potásico) pueden ser un complemento útil para reducir la calciuria y aumentar el volumen urinario89.
- Ortofosfato: puede administrarse en dosis de 30-40mg/kg/día90.
- Magnesio: es un conocido inhibidor de la mineralización y, además, reduce la absorción de oxalatos cuando se administra conjuntamente con los alimentos. La dosis recomendada91 es de 500mg/día/m2.
El empleo de análogos de la vitamina D puede tener un efecto adverso en estos pacientes, al incrementar la absorción de calcio y, como consecuencia de ello, la sobresaturación de oxalato cálcico92.
Puede necesitarse un tubo de gastrostomía en lactantes y niños pequeños para alcanzar estos objetivos55.
Dieta
La reducción de la ingesta de oxalatos es poco útil en la HOP, dado que la fuente endógena de oxalato prevalece; sin embargo, puede resultar más útil en los casos de hiperoxaluria entérica. Los alimentos más ricos en oxalato son: frutos secos, ciruelas, chocolate, té, Coca-Cola, remolacha, fresas, etc.
No debe restringirse la ingesta cálcica, ya que a consecuencia de ello aumenta la absorción intestinal de oxalato93.
Ha de evitarse la ingesta excesiva de vitamina C, especialmente en pacientes en diálisis, ya que el ácido ascórbico puede metabolizarse directamente a oxalato cálcico52.
Enzimas degradadoras de oxalato
Si bien los mamíferos no pueden metabolizar el oxalato, otros seres vivos tienen enzimas como la oxalato oxidasa y la oxalato decarboxilasa, capaces de degradarlo y que podrían proporcionar una solución novedosa para prevenir la acumulación de oxalato en la HOP94,95. En este sentido, la colonización intestinal con Oxalobacter formigenes, bacteria que metaboliza el oxalato, ha mostrado resultados prometedores en modelos de ratas con HOP y en algún estudio piloto en pacientes47,96.
Para pacientes con alteración de la flora intestinal el uso de probióticos (alimentos con microorganismos vivos añadidos que permanecen activos en el intestino) puede resultar beneficioso. Estas bacterias utilizan el oxalato intestinal como fuente de energía y previenen su absorción. Se ha empleado una combinación de lactobacilos productores de ácido láctico en pacientes con hiperoxaluria entérica, tanto inflamatoria como secundaria a resecciones intestinales, demostrándose una reducción de la oxaluria y de la saturación oxalocálcica97,98. En modelos de ratas con dietas altas en oxalato, suplementos de Oxalobacter formigenes redujeron de forma significativa la oxaluria. También se ha observado que el Oxalobacter formigenes interactúa con la mucosa colónica favoreciendo la excreción/secreción de oxalato de la sangre al intestino99.
Piridoxina
La piridoxina (vitamina B6) es, tal vez, la única medida capaz de reducir de forma eficaz la producción de oxalato, pero solo aplicable a los casos de HOP-I. El pyridoxal 5 fosfato es una forma de vitamina B6 que actúa como cofactor de la AGT aumentando la transaminación del glioxalato (precursor del oxalato) a glicina. La dosis recomendada es de 5mg/kg/día hasta un máximo de 20mg/kg/día100. Sin embargo, la seguridad de estas dosis no es bien conocida y se han descrito casos de neuropatía sensorial, por lo que es recomendable no pasar de 1g/día en adultos y corregir adecuadamente esta dosis para niños y lactantes. La respuesta se considera con un descenso de la oxaluria >30% después de un trimestre de tratamiento a máxima dosis2,3. Milliner et al.90 han comunicado la eficacia del tratamiento a largo plazo con ortofosfato y piridoxina, reduciendo la cristalización urinaria de oxalato cálcico en 25 pacientes con HOP y función renal conservada. El 75% de los pacientes se mantenían sin diálisis a los 20 años de tratamiento, habiendo caído la función renal a un promedio de 1,4ml/min/1,73m2 por año. Desde la perspectiva molecular, hay un subgrupo de pacientes portadores de una o dos copias de G170Arg y Phe152Ile, cuya mutación produce un destino anómalo de la AGT a la mitocondria (mistargeting) y que ha demostrado una mejor respuesta a la piridoxina101-104.
Manejo quirúrgico de la urolitiasis
El manejo de la litiasis en la HOP tiene como peculiaridad la presencia concomitante habitual de nefrocalcinosis. El tratamiento con litotricia conlleva el riesgo de aplicar ondas de choque a áreas de nefrocalcinosis. Por ello el manejo endoluminal endoscópico suele ser de elección, mientras que la cirugía abierta es excepcional en este campo105,106.
Diálisis
Una vez que la insuficiencia renal está establecida, todas las medidas generales mencionadas suelen ser ineficaces y debe planificarse el tratamiento sustitutivo renal. La hemodiálisis y más aún la diálisis peritoneal han demostrado ser ineficaces para depurar el oxalato generado en la HOP107,108. El oxalato es una molécula pequeña, fácilmente filtrable, pero la cantidad de oxalato producida por el hígado en la HOP suele ser significativamente mayor (350-600mg/dl diarios) que la capacidad de depuración de la diálisis convencional (80-180mg/dl en adultos), resultando en un depósito diario de oxalato cálcico de 180-360mg/día63,77,107-110. Idealmente, los niveles séricos deberían mantenerse por debajo de 30µMol/l62. De estos trabajos se desprende que, para conseguir el balance de oxalato en hemodiálisis, las sesiones deberían prolongarse de 13 a 15 horas, lo que resulta impracticable. Por lo tanto, la diálisis debe ser empleada mientras el paciente se encuentra a la espera de un trasplante, aplicando protocolos de diálisis de alta eficacia e intensiva7,111.
Trasplante renal
El empleo del trasplante renal aislado ha proporcionado resultados infaustos. Tras el implante, la recurrencia de la nefrocalcinosis es lo habitual, especialmente en casos de función renal retrasada o episodios de rechazo, por lo que debemos considerarlo como solución transitoria o de mantenimiento, mientras se planifica el trasplante hepático1,7,8,84,112,113. Con frecuencia el diagnóstico de HOP se ha realizado tras el trasplante renal, con la aparición de depósitos de oxalato y pérdida precoz del injerto por esta causa. Incluso estas situaciones se han documentado en casos desafortunados de donante vivo, donde no se había diagnosticado la HOP7.
Por lo tanto, dado el alto riesgo de desarrollo rápido de nefrocalcinosis, el trasplante renal aislado debería reservarse para formas más leves de HOP, con razonable buena respuesta a medidas conservadoras y vitamina B6. En estos casos, la insuficiencia renal aparece en edad más avanzada y progresa muy lentamente. El trasplante anticipado (en la etapa «prediálisis») es una decisión difícil, dada la incertidumbre de la velocidad de progresión del daño renal, siendo la mejor opción programarlo una vez iniciada la diálisis y tan precozmente como sea posible. La opción de un donante vivo está claramente desaconsejada ante el mal pronóstico del trasplante renal aislado, aunque puede considerarse ante situaciones excepcionales muy concretas.
La aparición de nefrocalcinosis en el injerto funcionante se debe a la rápida liberación de los depósitos sistémicos de oxalato cálcico. Por ello, es fundamental aplicar un protocolo agresivo pre y postrasplante inmediato, extremando el protocolo inmunosupresor, minimizando el tiempo de isquemia fría y manteniendo una diuresis fluida. Asimismo, debe suministrarse soporte dialítico precoz e intenso (diario) y añadir todas las medidas antilitogénicas descritas (piridoxina, inhibidores de la cristalización y diuréticos tiazídicos)7,114. Durante el seguimiento posterior es obligada la vigilancia periódica de la oxaluria y la potencial sobrecarga tisular de oxalato con medios diagnósticos de imagen.
Trasplante hepático y renal
Una vez que el diagnóstico de HOP es firme, el tratamiento prioritario y potencialmente curativo es el trasplante hepático7,115. Dado que lo habitual es estar ante un deterioro renal avanzado e irreversible, la indicación es el trasplante doble, hepático y renal1. El primer trasplante combinado hepático y renal efectuado con éxito para el tratamiento de la HOP fue publicado por Watts et al. en 1987116. Desde entonces un considerable número de publicaciones han puesto de manifiesto que el trasplante simultáneo hepático y renal es la opción terapéutica de elección en pacientes con daño renal avanzado, incluso en lactantes7,84,117-121. Datos obtenidos del Registro Europeo de Trasplante en HOP han demostrado una buena tasa de supervivencia del paciente: 80% a los 5 años y 68% a los 10 años119. Conviene resaltar que el trasplante debería programarse precozmente, con FG entre 15 y 30ml/min/1,73m2, para prevenir el depósito tisular de oxalatos.
También se han descrito otras alternativas de trasplante para situaciones especiales. La donación hepática parcial (split) de donante vivo conlleva riesgos al donante y la cantidad de hígado trasplantada puede ser insuficiente para prevenir la sobrecarga renal de oxalato del hígado remanente. Esta opción está actualmente desaconsejada, reservándose solamente para circunstancias excepcionales.
La alternativa de trasplante secuencial requiere dos donantes, pero es menos agresiva desde la perspectiva quirúrgica. Si el FG es mayor de 15-20ml/min, la opción es el hepático primero y evaluar la evolución renal posterior. Si el FG es <15-20ml/min, la opción es el trasplante renal primero. Esta última puede considerarse en casos de daño renal avanzado, pero cuando la progresión ha sido muy lenta, habitualmente en enfermos mayores de 50-60 años.
Trasplante hepático anticipado
La rápida progresión de la oxalosis una vez que la insuficiencia renal se ha establecido indica que el trasplante hepático debería realizarse previo al desarrollo de esta, y así evitar la necesidad del doble trasplante. El trasplante hepático anticipado, es decir, antes de que haya un daño renal irreversible, debería ser la opción de elección en casos severos, si disponemos de un diagnóstico certero y precoz7,122-126. Esta alternativa habría de aplicarse con una función renal entre 20 y 50ml/min115. Asimismo, basados en consideraciones previas sobre los niveles séricos de oxalato y su umbral de saturación, enfermos con oxalemias >50µmol/l si FG <20ml/min, o >20 µmol/l si FG <30ml/min, deberían ser considerados candidatos potenciales para trasplante hepático62,64.
En la tabla 3 se resumen las modalidades de trasplante en la HOP y se sugieren las indicaciones potenciales.
Tabla 3. Modalidades de trasplante en la hiperoxaluria primaria y potenciales indicaciones. 
FG: filtrado glomerular.
Perspectivas de terapia molecular
Los recientes avances en los mecanismos moleculares de la enfermedad han hecho posible el desarrollo de modelos genéticos animales y la experimentación in vivo de vías terapéuticas eficaces y no invasivas32,127.
Terapia génica
Enfermedades hereditarias como la HOP podrían ser resueltas proporcionando la secuencia normal del gen a las células defectuosas. El problema radicaría en que los hepatocitos no alcanzados siguen generando oxalato que sobrecarga el riñón. El desarrollo de vectores virales con alto tropismo hepático y baja respuesta inmune puede contribuir a que la terapia génica sea una opción realista para la HOP en el futuro128.
«Chaperones» químicos y terapia de regulación de la «proteostasis»
El término «proteostasis» se emplea para definir la homeostasis de proteínas intracelulares. Por lo tanto, combina conceptos de plegamiento y estabilidad proteica, degradación, tráfico intracelular y expresión de los sistemas de regulación proteica. Esta compleja y altamente regulada red determina el destino intracelular de las proteínas129. Estudios recientes han demostrado que pequeños componentes orgánicos, denominados «chaperones», pueden estar implicados en las alteraciones del plegamiento de la proteína, en el destino mitocondrial incorrecto (mistargeting) y en los procesos de agregación y degradación de la AGT23,78,130, dando lugar a variantes de presentación de la HOP.
Trasplante celular
El trasplante hepático parcial ha fallado en proporcionar una suficiente reducción en la producción de oxalato como para prevenir la HOP-I119. Sin embargo, el trasplante de células hepáticas es una prometedora alternativa que está siendo explorada en defectos genéticos del metabolismo hepático, incluyendo la HOP-I131,132. El trasplante de hepatocitos es un procedimiento mínimamente invasivo, aunque el número de hepatocitos sanos inyectados en una única sesión no es suficiente para corregir una elevada producción de oxalato por parte de los hepatocitos nativos. En nuestros experimentos con modelos de ratones con HOP-I, calculamos que aproximadamente un 40% de los hepatocitos necesitan ser transferidos para revertir el fenotipo hiperoxalúrico128.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
Agradecimientos
Un autor (A. Torres) es miembro de RedInRen (Red de Investigación Renal). Un autor (E Salido) es miembro de CIBERER (Centro de Investigación Biomedica en Red de Enfermedades Raras).
Referencias Bibliográficas
1. Cochat P, Rumsby G. Primary Hyperoxaluria. N Engl J Med 2013;369(7):649-58. [ Links ]
2. Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int 2009;75(12):1264-71. [ Links ]
3. Leumann E, Hoppe B. The primary hyperoxalurias. J Am Soc Nephrol 2001;12(9):1986-93. [ Links ]
4. Salido E, Pey AL, Rodriguez R, Lorenzo V. Primary hyperoxalurias: disorders of glyoxylate detoxification. Biochim Biophys Acta 2012;1822(9):1453-64. [ Links ]
5. van Woerden CS, Groothoff JW, Wanders RJ, Davin JC, Wijburg FA. Primary hyperoxaluria type 1 in The Netherlands: prevalence and outcome. Nephrol Dial Transplant 2003;18(2):273-9. [ Links ]
6. Cochat P, Deloraine A, Rotily M, Olive F, Liponski I, Deries N. Epidemiology of primary hyperoxaluria type 1. Societe de Nephrologie and the Societe de Nephrologie Pediatrique. Nephrol Dial Transplant 1995;10 Suppl 8:3-7. [ Links ]
7. Lorenzo V, Alvarez A, Torres A, Torregrosa V, Hernandez D, Salido E. Presentation and role of transplantation in adult patients with type 1 primary hyperoxaluria and the I244T AGXT mutation: Single-center experience. Kidney Int 2006;70(6):1115-9. [ Links ]
8. Harambat J, van Stralen KJ, Espinosa L, Groothoff JW, Hulton SA, Cerkauskiene R, et al. Characteristics and outcomes of children with primary oxalosis requiring renal replacement therapy. Clin J Am Soc Nephrol 2012;7(3):458-65. [ Links ]
9. Williams AW, Wilson DM. Dietary intake, absorption, metabolism, and excretion of oxalate. Semin Nephrol 1990;10(1):2-8. [ Links ]
10. Wanders RJ, Waterham HR. Peroxisomal disorders: the single peroxisomal enzyme deficiencies. Biochim Biophys Acta 2006;1763(12):1707-20. [ Links ]
11. Knight J, Jiang J, Assimos DG, Holmes RP. Hydroxyproline ingestion and urinary oxalate and glycolate excretion. Kidney Int 2006;70(11):1929-34. [ Links ]
12. Hoppe B. An update on primary hyperoxaluria. Nat Rev Nephrol 2012;8(8):467-75. [ Links ]
13. Hoppe B, Leumann E, von UG, Laube N, Hesse A. Diagnostic and therapeutic approaches in patients with secondary hyperoxaluria. Front Biosci 2003;8:e437-43. [ Links ]
14. Danpure CJ, Rumsby G. Molecular aetiology of primary hyperoxaluria and its implications for clinical management. Expert Rev Mol Med 2004;6(1):1-16. [ Links ]
15. Harambat J, Fargue S, Bacchetta J, Acquaviva C, Cochat P. Primary hyperoxaluria. Int J Nephrol 2011;2011:864580. [ Links ]
16. Monico CG, Rossetti S, Belostotsky R, Cogal AG, Herges RM, Seide BM, et al. Primary hyperoxaluria type III gene HOGA1 (formerly DHDPSL) as a possible risk factor for idiopathic calcium oxalate urolithiasis. Clin J Am Soc Nephrol 2011;6(9):2289-95. [ Links ]
17. Danpure CJ. Molecular aetiology of primary hyperoxaluria type 1. Nephron Exp Nephrol 2004;98(2):e39-44. [ Links ]
18. Danpure CJ. Molecular etiology of primary hyperoxaluria type 1: new directions for treatment. Am J Nephrol 2005;25(3):303-10. [ Links ]
19. Salido E, Pey AL, Rodriguez R, Lorenzo V. Primary hyperoxalurias: disorders of glyoxylate detoxification. Biochim Biophys Acta 2012;1822(9):1453-64. [ Links ]
20. Santana A, Torres A, Salido E. (Molecular pathology of primary hyperoxaluria). Nefrologia 2003;23 Suppl 1:90-7. [ Links ]
21. Cellini B, Bertoldi M, Montioli R, Paiardini A, Borri VC. Human wild-type alanine:glyoxylate aminotransferase and its naturally occurring G82E variant: functional properties and physiological implications. Biochem J 2007;408(1):39-50. [ Links ]
22. Cellini B, Montioli R, Voltattorni CB. Human liver peroxisomal alanine:glyoxylate aminotransferase: characterization of the two allelic forms and their pathogenic variants. Biochim Biophys Acta 2011;1814(11):1577-84. [ Links ]
23. Santana A, Salido E, Torres A, Shapiro LJ. Primary hyperoxaluria type 1 in the Canary Islands: a conformational disease due to I244T mutation in the P11L-containing alanine:glyoxylate aminotransferase. Proc Natl Acad Sci U S A 2003;100(12):7277-82. [ Links ]
24. Williams EL, Acquaviva C, Amoroso A, Chevalier F, Coulter-Mackie M, Monico CG, et al. Primary hyperoxaluria type 1: update and additional mutation analysis of the AGXT gene. Hum Mutat 2009;30(6):910-7. [ Links ]
25. Danpure CJ. Primary hyperoxaluria type 1: AGT mistargeting highlights the fundamental differences between the peroxisomal and mitochondrial protein import pathways. Biochim Biophys Acta 2006;1763(12):1776-84. [ Links ]
26. Danpure CJ, Cooper PJ, Wise PJ, Jennings PR. An enzyme trafficking defect in two patients with primary hyperoxaluria type 1: peroxisomal alanine/glyoxylate aminotransferase rerouted to mitochondria. J Cell Biol 1989;108(4):1345-52. [ Links ]
27. Purdue PE, Allsop J, Isaya G, Rosenberg LE, Danpure CJ. Mistargeting of peroxisomal L-alanine:glyoxylate aminotransferase to mitochondria in primary hyperoxaluria patients depends upon activation of a cryptic mitochondrial targeting sequence by a point mutation. Proc Natl Acad Sci U S A 1991;88(23):10900-4. [ Links ]
28. Danpure CJ, Purdue PE, Fryer P, Griffiths S, Allsop J, Lumb MJ, et al. Enzymological and mutational analysis of a complex primary hyperoxaluria type 1 phenotype involving alanine:glyoxylate aminotransferase peroxisome-to-mitochondrion mistargeting and intraperoxisomal aggregation. Am J Hum Genet 1993;53(2):417-32. [ Links ]
29. Williams EL, Kemper MJ, Rumsby G. A de novo mutation in the AGXT gene causing primary hyperoxaluria type 1. Am J Kidney Dis 2006;48(3):481-3. [ Links ]
30. Seargeant LE, deGroot GW, Dilling LA, Mallory CJ, Haworth JC. Primary oxaluria type 2 (L-glyceric aciduria): a rare cause of nephrolithiasis in children. J Pediatr 1991;118(6):912-4. [ Links ]
31. Dobbins JW, Binder HJ. Effect of bile salts and fatty acids on the colonic absorption of oxalate. Gastroenterology 1976;70(6):1096-100. [ Links ]
32. Bobrowski AE, Langman CB. Hyperoxaluria and systemic oxalosis: current therapy and future directions. Expert Opin Pharmacother 2006;7(14):1887-96. [ Links ]
33. Caspary WF. Enteric hyperoxaluria. N Engl J Med 1977;296(23):1357. [ Links ]
34. Caspary WF. Intestinal oxalate absorption. I. Absorption in vitro. Res Exp Med (Berl) 1977;171(1):13-24. [ Links ]
35. Dobbins JW, Binder HJ. Importance of the colon in enteric hyperoxaluria. N Engl J Med 1977;296(6):298-301. [ Links ]
36. Earnest DL. Enteric hyperoxaluria. Adv Intern Med 1979;24:407-27. [ Links ]
37. Diamond KL, Fox CC, Barch DH. Role of cecal pH in intestinal oxalate absorption in the rat. J Lab Clin Med 1988;112(3):352-6. [ Links ]
38. Canos HJ, Hogg GA, Jeffery JR. Oxalate nephropathy due to gastrointestinal disorders. Can Med Assoc J 1981;124(6):729-33. [ Links ]
39. Hofmann AF, Schmuck G, Scopinaro N, Laker MF, Sherr HP, Lorenzo D, et al. Hyperoxaluria associated with intestinal bypass surgery for morbid obesity: occurrence, pathogenesis and approaches to treatment. Int J Obes 1981;5(5):513-8. [ Links ]
40. Hofmann AF, Laker MF, Dharmsathaphorn K, Sherr HP, Lorenzo D. Complex pathogenesis of hyperoxaluria after jejunoileal bypass surgery. Oxalogenic substances in diet contribute to urinary oxalate. Gastroenterology 1983;84(2):293-300. [ Links ]
41. Lindsjo M, Danielson BG, Fellstrom B, Lithell H, Ljunghall S. Intestinal absorption of oxalate and calcium in patients with jejunoileal bypass. Scand J Urol Nephrol 1989;23(4):283-9. [ Links ]
42. Terribile M, Capuano M, Cangiano G, Carnovale V, Ferrara P, Petrarulo M, et al. Factors increasing the risk for stone formation in adult patients with cystic fibrosis. Nephrol Dial Transplant 2006;21(7):1870-5. [ Links ]
43. Cuvelier C, Goffin E, Cosyns JP, Wauthier M, de Strihou CY. Enteric hyperoxaluria: a hidden cause of early renal graft failure in two successive transplants: spontaneous late graft recovery. Am J Kidney Dis 2002;40(1):E3. [ Links ]
44. Mole DR, Tomson CR, Mortensen N, Winearls CG. Renal complications of jejuno-ileal bypass for obesity. QJM 2001;94(2):69-77. [ Links ]
45. Moller T, Muller G, Schutte W, Rogos R, Schneider W. (Oxalic acid resorption in patients with resection of the small intestine, jejunoileal bypass, Crohn disease and chronic pancreatitis). Dtsch Z Verdau Stoffwechselkr 1987;47(3):113-8. [ Links ]
46. Nelson WK, Houghton SG, Milliner DS, Lieske JC, Sarr MG. Enteric hyperoxaluria, nephrolithiasis, and oxalate nephropathy: potentially serious and unappreciated complications of Roux-en-Y gastric bypass. Surg Obes Relat Dis 2005;1(5):481-5. [ Links ]
47. Hoppe B, Beck B, Gatter N, von Unruh G, Tischer A, Hesse A, et al. Oxalobacter formigenes: a potential tool for the treatment of primary hyperoxaluria type 1. Kidney Int 2006;70(7):1305-11. [ Links ]
48. Kruse JA. Methanol and ethylene glycol intoxication. Crit Care Clin 2012;28(4):661-711. [ Links ]
49. Pomara C, Fiore C, D'Errico S, Riezzo I, Fineschi V. Calcium oxalate crystals in acute ethylene glycol poisoning: a confocal laser scanning microscope study in a fatal case. Clin Toxicol (Phila) 2008;46(4):322-4. [ Links ]
50. Samarneh MM, Shtaynberg N, Goldman M, Epstein E, Kleiner M, El-Sayegh S. Severe oxalosis with systemic manifestations. J Clin Med Res 2012;4(1):56-60. [ Links ]
51. Simpson GL, Ortwerth BJ. The non-oxidative degradation of ascorbic acid at physiological conditions. Biochim Biophys Acta 2000;1501(1):12-24. [ Links ]
52. Canavese C, Petrarulo M, Massarenti P, Berutti S, Fenoglio R, Pauletto D, et al. Long-term, low-dose, intravenous vitamin C leads to plasma calcium oxalate supersaturation in hemodialysis patients. Am J Kidney Dis 2005;45(3):540-9. [ Links ]
53. Wong K, Thomson C, Bailey RR, McDiarmid S, Gardner J. Acute oxalate nephropathy after a massive intravenous dose of vitamin C. Aust N Z J Med 1994;24(4):410-1. [ Links ]
54. Nasr SH, Kashtanova Y, Levchuk V, Markowitz GS. Secondary oxalosis due to excess vitamin C intake. Kidney Int 2006;70(10):1672. [ Links ]
55. Cochat P, Hulton SA, Acquaviva C, Danpure CJ, Daudon M, De Marchi M, et al. Primary hyperoxaluria type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant 2012;27(5):1729-36. [ Links ]
56. Skinner R, Tomson CR, Tapson JS. Long term survival on haemodialysis in primary hyperoxaluria. Int J Artif Organs 1990;13(7):412-5. [ Links ]
57. van Woerden CS, Groothoff JW, Wijburg FA, Waterham HR, Wanders RJ, Janssen MJ, et al. Primary hyperoxaluria remains undiagnosed in patients with hyperoxaluria and recurrent urolithiasis. Clin Chem 2007;53(8):1553-5. [ Links ]
58. Milliner DS, Wilson DM, Smith LH. Phenotypic expression of primary hyperoxaluria: comparative features of types I and II. Kidney Int 2001;59(1):31-6. [ Links ]
59. Belostotsky R, Seboun E, Idelson GH, Milliner DS, Becker-Cohen R, Rinat C, et al. Mutations in DHDPSL are responsible for primary hyperoxaluria type III. Am J Hum Genet 2010;87(3):392-9. [ Links ]
60. Cochat P, Fargue S, Bacchetta J, Bertholet-Thomas A, Sabot JF, Harambat J. (Primary hyperoxaluria). Nephrol Ther 2011;7(4):249-59. [ Links ]
61. Lorenz EC, Michet CJ, Milliner DS, Lieske JC. Update on oxalate crystal disease. Curr Rheumatol Rep 2013;15(7):340. [ Links ]
62. Hoppe B, Kemper MJ, Bokenkamp A, Portale AA, Cohn RA, Langman CB. Plasma calcium oxalate supersaturation in children with primary hyperoxaluria and end-stage renal failure. Kidney Int 1999;56(1):268-74. [ Links ]
63. Marangella M, Petrarulo M, Vitale C, Daniele PG, Sammartano S, Cosseddu D, et al. Serum calcium oxalate saturation in patients on maintenance haemodialysis for primary hyperoxaluria or oxalosis-unrelated renal diseases. Clin Sci (Lond) 1991;81(4):483-90. [ Links ]
64. Marangella M, Cosseddu D, Petrarulo M, Vitale C, Linari F. Thresholds of serum calcium oxalate supersaturation in relation to renal function in patients with or without primary hyperoxaluria. Nephrol Dial Transplant 1993;8(12):1333-7. [ Links ]
65. Lorenzo V, Torres A, Hernandez D. Evolución de la enfermedad ósea en pacientes con hiperoxaluria primaria. Nefrologia 1990;1:53-60. [ Links ]
66. Baethge BA, Sanusi ID, Landreneau MD, Rohr MS, McDonald JC. Livedo reticularis and peripheral gangrene associated with primary hyperoxaluria. Arthritis Rheum 1988;31(9):1199-203. [ Links ]
67. Gherardi G, Poggi A, Sisca S, Calderaro V, Bonucci E. Bone oxalosis and renal osteodystrophy. Arch Pathol Lab Med 1980;104(2):105-11. [ Links ]
68. Julian BA, Faugere MC, Malluche HH. Oxalosis in bone causing a radiographical mimicry of renal osteodystrophy. Am J Kidney Dis 1987;9(5):436-40. [ Links ]
69. Lorenzo V, Hernandez D, Dominguez M, Rodriguez A, Torres A. Oxalosis as a cause of absolute resistance to rHuEpo in chronic haemodialysis patients. Nephrol Dial Transplant 1992;7(11):1163-4. [ Links ]
70. Sahin G, Acikalin MF, Yalcin AU. Erythropoietin resistance as a result of oxalosis in bone marrow. Clin Nephrol 2005;63(5):402-4. [ Links ]
71. Daudon M, Jungers P, Bazin D. Peculiar morphology of stones in primary hyperoxaluria. N Engl J Med 2008;359(1):100-2. [ Links ]
72. Rumsby G. An overview of the role of genotyping in the diagnosis of the primary hyperoxalurias. Urol Res 2005;33(5):318-20. [ Links ]
73. Milliner DS. The primary hyperoxalurias: an algorithm for diagnosis. Am J Nephrol 2005;25(2):154-60. [ Links ]
74. Marangella M, Petrarulo M, Bianco O, Vitale C, Finocchiaro P, Linari F. Glycolate determination detects type I primary hyperoxaluria in dialysis patients. Kidney Int 1991;39(1):149-54. [ Links ]
75. Dietzen DJ, Wilhite TR, Kenagy DN, Milliner DS, Smith CH, Landt M. Extraction of glyceric and glycolic acids from urine with tetrahydrofuran: utility in detection of primary hyperoxaluria. Clin Chem 1997;43(8 Pt 1):1315-20. [ Links ]
76. Barratt TM, Kasidas GP, Murdoch I, Rose GA. Urinary oxalate and glycolate excretion and plasma oxalate concentration. Arch Dis Child 1991;66(4):501-3. [ Links ]
77. Marangella M, Petrarulo M, Cosseddu D, Vitale C, Linari F. Oxalate balance studies in patients on hemodialysis for type I primary hyperoxaluria. Am J Kidney Dis 1992;19(6):546-53. [ Links ]
78. Pey AL, Salido E, Sanchez-Ruiz JM. Role of low native state kinetic stability and interaction of partially unfolded states with molecular chaperones in the mitochondrial protein mistargeting associated with primary hyperoxaluria. Amino Acids 2011;41(5):1233-45. [ Links ]
79. Purdue PE, Lumb MJ, Fox M, Griffo G, Hamon-Benais C, Povey S, et al. Characterization and chromosomal mapping of a genomic clone encoding human alanine:glyoxylate aminotransferase. Genomics 1991;10(1):34-42. [ Links ]
80. Takada Y, Kaneko N, Esumi H, Purdue PE, Danpure CJ. Human peroxisomal L-alanine: glyoxylate aminotransferase. Evolutionary loss of a mitochondrial targeting signal by point mutation of the initiation codon. Biochem J 1990;268(2):517-20. [ Links ]
81. Rumsby G, Weir T, Samuell CT. A semiautomated alanine:glyoxylate aminotransferase assay for the tissue diagnosis of primary hyperoxaluria type 1. Ann Clin Biochem 1997;34(Pt 4):400-4. [ Links ]
82. Danpure CJ, Jennings PR, Fryer P, Purdue PE, Allsop J. Primary hyperoxaluria type 1: genotypic and phenotypic heterogeneity. J Inherit Metab Dis 1994;17(4):487-99. [ Links ]
83. Rumsby G. Experience in prenatal diagnosis of primary hyperoxaluria type 1. J Nephrol 1998;11 Suppl 1:13-4. [ Links ]
84. Scheinman JI. Primary hyperoxaluria: therapeutic strategies for the 90's. Kidney Int 1991;40(3):389-99. [ Links ]
85. Borghi L, Meschi T, Amato F, Briganti A, Novarini A, Giannini A. Urinary volume, water and recurrences in idiopathic calcium nephrolithiasis: a 5-year randomized prospective study. J Urol 1996;155(3):839-43. [ Links ]
86. Leumann E, Hoppe B, Neuhaus T. Management of primary hyperoxaluria: efficacy of oral citrate administration. Pediatr Nephrol 1993;7(2):207-11. [ Links ]
87. Rodgers A, Allie-Hamdulay S, Jackson G. Therapeutic action of citrate in urolithiasis explained by chemical speciation: increase in pH is the determinant factor. Nephrol Dial Transplant 2006;21(2):361-9. [ Links ]
88. Marangella M, Bagnis C, Bruno M, Vitale C, Petrarulo M, Ramello A. Crystallization inhibitors in the pathophysiology and treatment of nephrolithiasis. Urol Int 2004;72 Suppl 1:6-10. [ Links ]
89. Pak CY, Heller HJ, Pearle MS, Odvina CV, Poindexter JR, Peterson RD. Prevention of stone formation and bone loss in absorptive hypercalciuria by combined dietary and pharmacological interventions. J Urol 2003;169(2):465-9. [ Links ]
90. Milliner DS, Eickholt JT, Bergstralh EJ, Wilson DM, Smith LH. Results of long-term treatment with orthophosphate and pyridoxine in patients with primary hyperoxaluria. N Engl J Med 1994;331(23):1553-8. [ Links ]
91. Zimmermann DJ, Voss S, von Unruh GE, Hesse A. Importance of magnesium in absorption and excretion of oxalate. Urol Int 2005;74(3):262-7. [ Links ]
92. Marangella M, Vitale C, Cosseddu D, Petrarulo M, Linari F. Effects of oral and intravenous calcitriol on serum calcium oxalate saturation in dialysis patients. Clin Sci (Lond) 1993;85(3):309-14. [ Links ]
93. von Unruh GE, Voss S, Sauerbruch T, Hesse A. Dependence of oxalate absorption on the daily calcium intake. J Am Soc Nephrol 2004;15(6):1567-73. [ Links ]
94. Hatch M, Freel RW. The roles and mechanisms of intestinal oxalate transport in oxalate homeostasis. Semin Nephrol 2008;28(2):143-51. [ Links ]
95. Grujic D, Salido EC, Shenoy BC, Langman CB, McGrath ME, Patel RJ, et al. Hyperoxaluria is reduced and nephrocalcinosis prevented with an oxalate-degrading enzyme in mice with hyperoxaluria. Am J Nephrol 2009;29(2):86-93. [ Links ]
96. Hoppe B, Groothoff JW, Hulton SA, Cochat P, Niaudet P, Kemper MJ, et al. Efficacy and safety of Oxalobacter formigenes to reduce urinary oxalate in primary hyperoxaluria. Nephrol Dial Transplant 2011;26(11):3609-15. [ Links ]
97. Lieske JC, Goldfarb DS, De Simone SC, Regnier C. Use of a probiotic to decrease enteric hyperoxaluria. Kidney Int 2005;68(3):1244-9. [ Links ]
98. Campieri C, Campieri M, Bertuzzi V, Swennen E, Matteuzzi D, Stefoni S, et al. Reduction of oxaluria after an oral course of lactic acid bacteria at high concentration. Kidney Int 2001;60(3):1097-105. [ Links ]
99. Hatch M, Cornelius J, Allison M, Sidhu H, Peck A, Freel RW. Oxalobacter sp. reduces urinary oxalate excretion by promoting enteric oxalate secretion. Kidney Int 2006;69(4):691-8. [ Links ]
100. Hoppe B, Latta K, von Schnakenburg C, Kemper MJ. Primary hyperoxaluria--the German experience. Am J Nephrol 2005;25(3):276-81. [ Links ]
101. Harambat J, Fargue S, Acquaviva C, Gagnadoux MF, Janssen F, Liutkus A, et al. Genotype-phenotype correlation in primary hyperoxaluria type 1: the p.Gly170Arg AGXT mutation is associated with a better outcome. Kidney Int 2010;77(5):443-9. [ Links ]
102. Monico CG, Rossetti S, Olson JB, Milliner DS. Pyridoxine effect in type I primary hyperoxaluria is associated with the most common mutant allele. Kidney Int 2005;67(5):1704-9. [ Links ]
103. Monico CG, Olson JB, Milliner DS. Implications of genotype and enzyme phenotype in pyridoxine response of patients with type I primary hyperoxaluria. Am J Nephrol 2005;25(2):183-8. [ Links ]
104. van Woerden CS, Groothoff JW, Wijburg FA, Annink C, Wanders RJ, Waterham HR. Clinical implications of mutation analysis in primary hyperoxaluria type 1. Kidney Int 2004;66(2):746-52. [ Links ]
105. Straub M, Gschwend J, Zorn C. Pediatric urolithiasis: the current surgical management. Pediatr Nephrol 2010;25(7):1239-44. [ Links ]
106. Kerbl K, Clayman RV. Endourologic treatment of nephrocalcinosis. Urology 2000;56(3):508. [ Links ]
107. Illies F, Bonzel KE, Wingen AM, Latta K, Hoyer PF. Clearance and removal of oxalate in children on intensified dialysis for primary hyperoxaluria type 1. Kidney Int 2006;70(9):1642-8. [ Links ]
108. Watts RW, Veall N, Purkiss P. Oxalate dynamics and removal rates during haemodialysis and peritoneal dialysis in patients with primary hyperoxaluria and severe renal failure. Clin Sci (Lond) 1984;66(5):591-7. [ Links ]
109. Marangella M, Bagnis C, Bruno M, Petrarulo M, Gabella P, Linari F. Determinants of oxalate balance in patients on chronic peritoneal dialysis. Am J Kidney Dis 1993;21(4):419-26. [ Links ]
110. Yamauchi T, Quillard M, Takahashi S, Nguyen-Khoa M. Oxalate removal by daily dialysis in a patient with primary hyperoxaluria type 1. Nephrol Dial Transplant 2001;16(12):2407-11. [ Links ]
111. Cochat P, Liutkus A, Fargue S, Basmaison O, Ranchin B, Rolland MO. Primary hyperoxaluria type 1: still challenging! Pediatr Nephrol 2006;21(8):1075-81. [ Links ]
112. Cibrik DM, Kaplan B, Arndorfer JA, Meier-Kriesche HU. Renal allograft survival in patients with oxalosis. Transplantation 2002;74(5):707-10. [ Links ]
113. Bergstralh EJ, Monico CG, Lieske JC, Herges RM, Langman CB, Hoppe B, et al. Transplantation outcomes in primary hyperoxaluria. Am J Transplant 2010;10(11):2493-501. [ Links ]
114. Scheinman JI, Najarian JS, Mauer SM. Successful strategies for renal transplantation in primary oxalosis. Kidney Int 1984;25(5):804-11. [ Links ]
115. Cochat P, Fargue S, Harambat J. Primary hyperoxaluria type 1: strategy for organ transplantation. Curr Opin Organ Transplant 2010;15(5):590-3. [ Links ]
116. Watts RW, Morgan SH, Danpure CJ, Purkiss P, Calne RY, Rolles K, et al. Combined hepatic and renal transplantation in primary hyperoxaluria type I: clinical report of nine cases. Am J Med 1991;90(2):179-88. [ Links ]
117. Cibrik DM, Kaplan B, Arndorfer JA, Meier-Kriesche HU. Renal allograft survival in patients with oxalosis. Transplantation 2002;74(5):707-10. [ Links ]
118. Harps E, Brinkert F, Ganschow R, Briem-Richter A, van Husen M, Schmidtke S, et al. Immediate postoperative intensive care treatment of pediatric combined liver-kidney transplantation: outcome and prognostic factors. Transplantation 2011;91(10):1127-31. [ Links ]
119. Jamieson NV. A 20-year experience of combined liver/kidney transplantation for primary hyperoxaluria (PH1): the European PH1 transplant registry experience 1984-2004. Am J Nephrol 2005;25(3):282-9. [ Links ]
120. Mehrabi A, Fonouni H, Ayoub E, Rahbari NN, Müller SA, Morath Ch, et al. A single center experience of combined liver kidney transplantation. Clin Transplant 2009;23 Suppl 21:102-14. [ Links ]
121. Monico CG, Milliner DS. Combined liver-kidney and kidney-alone transplantation in primary hyperoxaluria. Liver Transpl 2001;7(11):954-63. [ Links ]
122. Kemper MJ, Nolkemper D, Rogiers X, Timmermann K, Sturm E, Malago M, et al. Preemptive liver transplantation in primary hyperoxaluria type 1: timing and preliminary results. J Nephrol 1998;11 Suppl 1:46-8. [ Links ]
123. Cochat P, Faure JL, Divry P, Danpure CJ, Descos B, Wright C, et al. Liver transplantation in primary hyperoxaluria type 1. Lancet 1989;1(8647):1142-3. [ Links ]
124. Perera MT, Sharif K, Lloyd C, Foster K, Hulton SA, Mirza DF, et al. Pre-emptive liver transplantation for primary hyperoxaluria (PH-I) arrests long-term renal function deterioration. Nephrol Dial Transplant 2011;26(1):354-9. [ Links ]
125. Brinkert F, Ganschow R, Helmke K, Harps E, Fischer L, Nashan B, et al. Transplantation procedures in children with primary hyperoxaluria type 1: outcome and longitudinal growth. Transplantation 2009;87(9):1415-21. [ Links ]
126. Cochat P, Groothoff J. Primary hyperoxaluria type 1: practical and ethical issues. Pediatr Nephrol 2013;28(12):2273-81. [ Links ]
127. Salido EC, Li XM, Lu Y, Wang X, Santana A, Roy-Chowdhury N, et al. Alanine-glyoxylate aminotransferase-deficient mice, a model for primary hyperoxaluria that responds to adenoviral gene transfer. Proc Natl Acad Sci U S A 2006;103(48):18249-54. [ Links ]
128. Salido E, Rodriguez-Pena M, Santana A, Beattie SG, Petry H, Torres A. Phenotypic correction of a mouse model for primary hyperoxaluria with adeno-associated virus gene transfer. Mol Ther 2011;19(5):870-5. [ Links ]
129. Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem 2009;78:959-91. [ Links ]
130. Albert A, Yunta C, Arranz R, Peña A, Salido E, Valpuesta JM, et al. Structure of GroEL in complex with an early folding intermediate of alanine glyoxylate aminotransferase. J Biol Chem 2010;285(9):6371-6. [ Links ]
131. Guha C, Yamanouchi K, Jiang J, Wang X, Roy Chowdhury N, Santana A, et al. Feasibility of hepatocyte transplantation-based therapies for primary hyperoxalurias. Am J Nephrol 2005;25(2):161-70. [ Links ]
132. Jiang J, Salido EC, Guha C, Wang X, Moitra R, Liu L, et al. Correction of hyperoxaluria by liver repopulation with hepatocytes in a mouse model of primary hyperoxaluria type-1. Transplantation 2008;85(9):1253-60. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Víctor Lorenzo,
Servicio de Nefrología, Hospital Universitario de Canarias,
Llombet 27,
38296, San Cristóbal de La Laguna,
Santa Cruz de Tenerife
E-mail: vls243@gmail.com
Enviado a Revisar: 8 Nov. 2013
Aceptado el: 20 Ene. 2014

