










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNefrología (Madrid)
versión On-line ISSN 1989-2284versión impresa ISSN 0211-6995
Nefrología (Madr.) vol.34 no.4 Cantabria 2014
https://dx.doi.org/10.3265/Nefrologia.pre2014.Apr.12381
Inhibición de mTOR, proteínas Akt y enfermedad renal crónica
mTOR inhibition, Akt proteins and chronic kidney disease
Eva Márquez y Julio Pascual
Servicio de Nefrología. Hospital del Mar
Institut Mar d'Investigacions Mediques
Red Temática de Investigación Renal (RedinRen). Barcelona
Dirección para correspondencia
Apesar de que hasta el 9% de la población presenta algún grado de enfermedad renal crónica (ERC), siguen sin conocerse en detalle las vías fisiopatológicas que participan en su progresión1. Los podocitos, células glomerulares epiteliales altamente especializadas, tienen un papel fundamental en el mantenimiento de la barrera de filtración glomerular. Para este mantenimiento es necesaria una regulación precisa de su citoesqueleto de actina; su reorganización produce alteraciones morfológicas y funcionales que causan la aparición de proteinuria. La familia de proteínas Akt son serina/treonina kinasas que regulan funciones metabólicas, de crecimiento y de supervivencia celular2 (figura 1). En podocitos está descrito su papel protector frente a la apoptosis3-6. La nefrina y la CD2AP, imprescindibles para el mantenimiento de la estructura y funcionalidad celular, tienen un papel en la regulación de la apoptosis celular7 y una posible función en la regulación del citoesqueleto8, ambos efectos vía Akt.
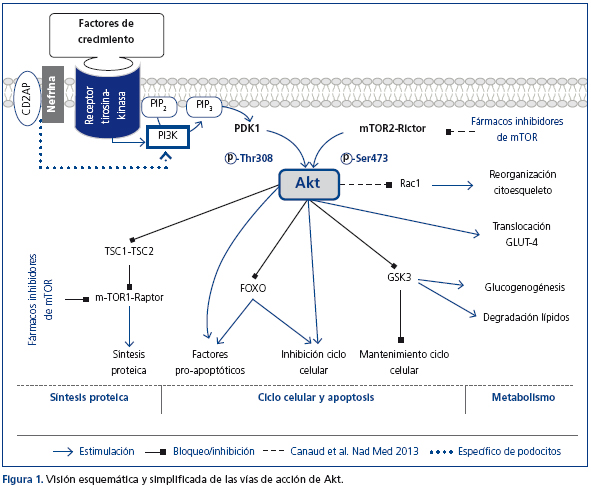
Recientemente Canaud et al. han publicado los resultados de un estudio en el que, utilizando modelos experimentales in vivo, in vitro y biopsias humanas, demuestran la activación de Akt2 como mecanismo protector en el podocito frente a una reducción de la masa renal9. La pérdida de Akt2 o la disminución en una de las fosforilaciones necesarias para su actividad [pAkt (Ser473)] agrava la lesión podocitaria, propiciando la aparición de proteinuria. El fallo en su fosforilación bloqueando el complejo mTOR2 mediante el uso de sirolimus podría explicar, al menos en parte, los efectos indeseados de este grupo de fármacos observados en algunos pacientes trasplantados.
Este estudio destaca metodológicamente por el uso de animales modificados genéticamente. Sus autores generan animales knock-out (KO) totales para Akt2 y KO-específico de podocitos, pudiendo valorarse así la trascendencia de estas células en la evolución de las lesiones renales. Estos animales, junto con sus respectivos y adecuados controles, son sometidos a una reducción de la masa renal mediante una nefrectomía subtotal o evaluados a los 13 meses de edad como modelo de envejecimiento. Del mismo modo, desarrollan un ratón KO-específico para Rictor, componente esencial del complejo mTOR2.
En situación de reducción de la masa renal se observa un incremento a nivel proteico de Akt2, que está presente fundamentalmente a nivel podocitario. Su aumento parece ser un mecanismo protector ante el daño, ya que, en relación con sus respectivos controles, los animales KO para Akt2 muestran lesión podocitaria con borramiento pedicelar asociada a un incremento en Rac1, incremento en la apoptosis a nivel glomerular, mayor grado de lesión glomerular, y en consecuencia mayor nivel de albuminuria. Alteraciones similares se encuentran en los ratones KO para Akt2 específicamente en los podocitos, confirmando la trascendencia de Akt2 podocitario en el mantenimiento de la función renal. Pero no solo la ausencia de Akt2 causa alteraciones renales: un déficit su fosforilación, como el observado en los ratones KO para Rictor, causa alteraciones equivalentes. Los resultados in vitro refuerzan los hallazgos in vivo. Los podocitos KO para Akt2 o tratados con sirolimus muestran alteración de su citoesqueleto con redistribución de las fibras de actina y aparición de focos de adhesión.
Los estudios realizados en biopsias humanas muestran, al igual que en el modelo animal, que Akt2 se expresa fundamentalmente en podocitos. Además se observa un aumento de pAkt (Ser473) a nivel glomerular en pacientes con diferentes patologías, principalmente de origen vascular.
El estudio de pacientes trasplantados con diferentes grados de disfunción renal muestra que solo los pacientes con marcada reducción de la masa renal (severe nephron reduction) presentan proteinuria realizando tratamiento con sirolimus. Los pacientes con peor función renal muestran tinciones intensas para pAkt (Ser473) y Rictor, lo cual no se encuentra en los pacientes con peor función renal y tratados con sirolimus. Este grupo evidencia mayor apoptosis celular a nivel glomerular y clínicamente presencia de proteinuria. Uno de los hallazgos más destacables es la correlación temporal entre la retirada del sirolimus, el aumento de pAkt (Ser473) en la biopsia renal y la disminución de la proteinuria.
Con un excelente diseño en los estudios in vivo e in vitro, este trabajo identifica Akt2 como elemento central en la fisiopatología de la lesión podocitaria en la ERC. Los modelos utilizados son de reducción de la masa renal, por lo que los hallazgos pueden ser potencialmente aplicables a la ERC de cualquier etiología. Recientemente se ha demostrado que la activación de Rac1 causa alteraciones del citoesqueleto del podocito, llevando al borramiento de los pedicelos10. Canaud et al. demuestran que la disminución de Akt2 activa Rac1 causando alteraciones del citoesqueleto.
De este modo se confirman estudios previos en podocitos y en otros modelos experimentales que demuestran que los fármacos inhibidores de mTOR no actúan únicamente bloqueando mTOR1, sino también mTOR211,12. Además, del trabajo de Canaud et al. puede concluirse que existe otra kinasa, no solo mTOR2, que fosforila Akt, ya que en modelo KO para Rictor no presenta un ausencia total de pAkt (Ser473). El análisis de Akt se realiza mediante estudios de Akt total, sus isoformas y pAkt (Ser473), pero en ningún modelo estudian el estado de fosforilación de Akt2 específicamente ni pAkt(Thr308), que, aunque no sea dependiente del complejo mTOR2, aportaría información relevante para la evaluación completa de la situación funcional de Akt13. Por otro lado, la valoración de Rictor-mTOR2 en los diferentes modelos animales podría contribuir a aclarar su papel y regulación. Sorprendentemente, en presencia de sirolimus, no detectan modificaciones en las moléculas fosforiladas por el complejo mTOR1, el cual no estudian específicamente.
Uno de los hallazgos más destacables es que el defecto en la fosforilación de Akt parece explicar, al menos en parte, el desarrollo de proteinuria observado en los pacientes trasplantados con mala función renal cuando se introduce un inhibidor de mTOR14. Los pacientes se agrupan en elevado y bajo filtrado glomerular estimado, pero no se explican la función renal y proteinuria previa a la introducción de sirolimus o la duración del tratamiento con sirolimus, datos que ayudarían a establecer el posible papel predictor de Akt sobre la función renal tras la introducción del tratamiento. Los estudios en biopsias renales humanas muestran un incremento en pAkt (Ser473) glomerular en diferentes patologías, pero en ninguno de los grupos estudian específicamente la isoforma Akt2, lo que obviamente resultaría de interés en el contexto del estudio presentado.
Futuras investigaciones, en modelos tanto animales como celulares, deberían dirigirse a aclarar cuáles son los estímulos que causan la elevación de Akt2, los cuales podrían ser específicos de diferentes patologías. Este estudio referido se centra en modelos de daño renal establecido, pero el estudio de esta vía en modelos con daño precoz resultaría trascendente. La prevalencia de la nefropatía diabética y el papel de la vía PI3K/Akt en su fisiopatología, incluida la lesión podocitaria3-6, la convierten en un evidente objetivo para profundizar en el análisis del papel de Akt2. Asimismo, las investigaciones en profundidad de mecanismos de apoptosis, regulación del ciclo celular (especialmente en el podocito, célula terminalmente diferenciada) y modificaciones del citoesqueleto, todo ello regulado por Akt, son necesarias para comprender las consecuencias fisiopatológicas de los cambios en esta molécula.
La profundización en estos diferentes campos facilitaría poder definir posibles dianas terapéuticas que llevasen al diseño de nuevos fármacos o a la utilización de los actuales para conseguir frenar o incluso prevenir el desarrollo de un daño renal establecido.
Finalmente, los hallazgos en las biopsias humanas abren la puerta a nuevas herramientas de decisión clínica en el manejo de la inmunosupresión. Con estudios diseñados específicamente se podría confirmar el probable valor pronóstico de Akt total, sus fosforilaciones o sus isoformas a la hora de valorar biopsias postrasplante como paso previo a una conversión a inhibidores de mTOR. El estudio de posibles marcadores no invasivos relacionados con los resultados de este estudio supondría una mejora en la práctica clínica habitual.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
Referencias Bibliográficas
1. Otero A, de Francisco A, Gayoso P, Garcia F, Group ES. Prevalence of chronic renal disease in Spain: results of the EPIRCE study. Nefrologia 2010;30(1):78-86. [ Links ]
2. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007;129(7):1261-74. [ Links ]
3. Wang XM, Yao M, Liu SX, Hao J, Liu QJ, Gao F. Interplay between the Notch and PI3K/Akt pathways in high glucose-induced podocyte apoptosis. Am J Physiol Renal Physiol 2014;306:F205-13. [ Links ]
4. Tejada T, Catanuto P, Ijaz A, Santos JV, Xia X, Sanchez P, et al. Failure to phosphorylate AKT in podocytes from mice with early diabetic nephropathy promotes cell death. Kidney Int 2008;73(12):1385-93. [ Links ]
5. Bussolati B, Deregibus MC, Fonsato V, Doublier S, Spatola T, Procida S, et al. Statins prevent oxidized LDL-induced injury of glomerular podocytes by activating the phosphatidylinositol 3-kinase/AKT-signaling pathway. J Am Soc Nephrol 2005;16(7):1936-47. [ Links ]
6. Logar CM, Brinkkoetter PT, Krofft RD, Pippin JW, Shankland SJ. Darbepoetin alfa protects podocytes from apoptosis in vitro and in vivo. Kidney Int 2007;72(4):489-98. [ Links ]
7. Huber TB, Hartleben B, Kim J, Schmidts M, Schermer B, Keil A, et al. Nephrin and CD2AP associate with phosphoinositide 3-OH kinase and stimulate AKT-dependent signaling. Mol Cell Biol 2003;23(14):4917-28. [ Links ]
8. Zhu J, Sun N, Aoudjit L, Li H, Kawachi H, Lemay S, et al. Nephrin mediates actin reorganization via phosphoinositide 3-kinase in podocytes. Kidney Int 2008;73(5):556-66. [ Links ]
9. Canaud G, Bienaime F, Viau A, Treins C, Baron W, Nguyen C, et al. AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat Med 2013;19(10):1288-96. [ Links ]
10. Yu H, Suleiman H, Kim AH, Miner JH, Dani A, Shaw AS, et al. Rac1 activation in podocytes induces rapid foot process effacement and proteinuria. Mol Cell Biol 2013;33(23):4755-64. [ Links ]
11. Vollenbroker B, George B, Wolfgart M, Saleem MA, Pavenstadt H, Weide T. mTOR regulates expression of slit diaphragm proteins and cytoskeleton structure in podocytes. Am J Physiol Renal Physiol 2009;296(2):F418-26. [ Links ]
12. Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012;335(6076):1638-43. [ Links ]
13. Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med 2005;9(1):59-71. [ Links ]
14. Diekmann F, Budde K, Oppenheimer F, Fritsche L, Neumayer HH, Campistol JM. Predictors of success in conversion from calcineurin inhibitor to sirolimus in chronic allograft dysfunction. Am J Transplant 2004;4(11):1869-75. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Julio Pascual
Servicio de Nefrología
Hospital del Mar
Institut Mar d'Investigacions Mediques
Red Temática de Investigación Renal (RedinRen)
08003, Barcelona
E-mail: julpascual@gmail.com
Enviado a Revisar: 30 Nov. 2013
Aceptado el: 21 Abr. 2014

