










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versión impresa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.81 no.10 oct. 2006
COMUNICACIÓN CORTA
Recidiva de tumor neuroectodérmico primitivo periférico orbitario con metástasis sistémica
Recurrence of peripheral primitive neuroectodermal tumor of the orbit with systemic metastases
Romero R. 1 , Abelairas J. 2 , Sanz J. 1 , Ruiz M.M. 1 , Sendagorta E. 1
Sección de Oftalmología Infantil. Hospital Universitario La Paz. Madrid. España.
1 Licenciado en Medicina
2 Doctor en Medicina
Comunicación presentada como comunicación oral en el LXXX Congreso de la S.E.O. (Córdoba 2004)
Dirección para correspondencia
RESUMEN
Caso clínico:Paciente varón de 6 años con proptosis del ojo derecho. Las pruebas de imagen detectaron una masa en la pared medial de la órbita derecha que fue biopsiada; el diagnóstico histopatológico fue de tumor neuroectodérmico primitivo. El paciente recibió tratamiento quimioterápico. Tras 7 años en remisión el paciente presentó recidiva del tumor orbitario con metástasis sistémicas que fue tratada sin éxito con quimioterapia, radioterapia, y exenteración orbitaria.
Discusión: La localización orbitaria de estos tumores es extremadamente rara. El diagnóstico diferencial debe realizarse con otros tumores de células pequeñas redondeadas, siendo fundamentales las técnicas inmunohistoquímicas y/o ultraestructurales.
Palabras clave: Tumor neuroectodérmico primitivo periférico, recidiva, órbita, proptosis, tumor de células pequeñas redondeadas.
ABSTRACT
Case report: A six-year-old boy presented with proptosis of the right eye. Imaging studies detected a mass in the medial wall of the right orbit. This mass was biopsied revealing a histopathologic diagnosis of primitive neuroectodermal tumor, so chemotherapy treatment was given. After seven years in remission he presented with a recurrence of the orbital tumor and was found to also have systemic metastases. Treatment with chemotherapy, radiotherapy and orbital exenteration was unsuccessful.
Discussion: The orbital occurrence of these tumors is extremely rare. Differentiation from other small round cell tumors requires immunohistochemical and ultrastructural techniques (Arch Soc Esp Oftalmol 2006; 81: 599-602).
Key words: Peripheral primitive neuroectodermal tumor, recurrence, orbit, proptosis, small round cell tumors.
Introducción
Los tumores neuroectodérmicos primitivos periféricos (PPNET) son tumores malignos de tejidos blandos que presumiblemente tienen su origen en la cresta neural y aparecen fuera del sistema nervioso central y del sistema nervioso simpático. Representan el 4-17% de los tumores de tejidos blandos en la edad pediátrica.
La localización orbitaria de estos tumores es extremadamente rara. En nuestro conocimiento solo existen diez casos publicados en la literatura (1-5), ocho de ellos en la edad pediátrica.
Caso Clínico
Paciente varón de seis años de edad sin antecedentes personales ni familiares de interés que acude al servicio de urgencias en enero de 1997 por presentar exoftalmos unilateral derecho de tres días de evolución. La exploración reveló limitación de la supraducción del ojo derecho (OD) con retropulsión disminuida y exoftalmos levemente doloroso de dicho ojo (exoftalmometría ojo derecho: 16mm y ojo izquierdo (OI): 11 mm). El resto de la exploración oftalmológica fue normal.
La tomografía axial computerizada (TAC) y la resonancia magnética nuclear (RMN) mostraron una masa extraconal (28 mm ¥ 18 mm ¥ 15 mm) de aspecto compacto, situada en la pared interna de la órbita derecha que desplazaba el globo ocular inferotemporalmente, y que captaba contraste (figs. 1 y 2). No se apreció invasión de estructuras vecinas.
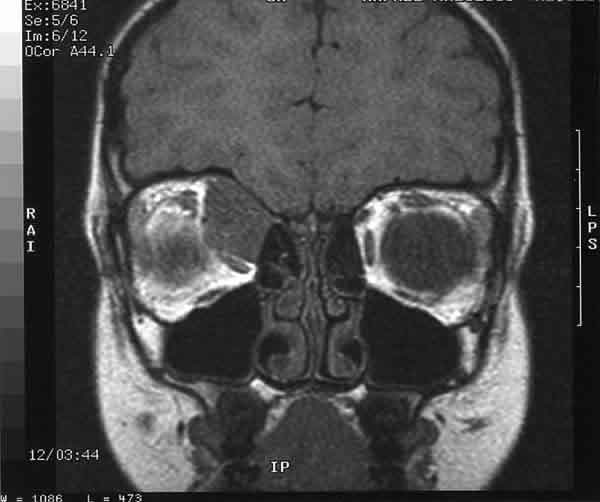
Fig. 1.RMN en T1, corte coronal.
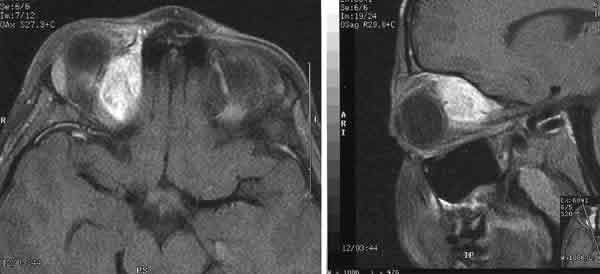
Fig. 2.RMN con contraste (Gadolinio) en T1, corte axial (izquierda) y corte sagital (derecha).
Bajo anestesia general se realizó una orbitotomía transconjuntival resecándose parcialmente el tumor. El estudio histopatológico reveló un tumor maligno de células pequeñas redondeadas con frecuentes figuras de mitosis (fig. 3); no se objetivaron rosetas ni pseudorosetas. Con técnicas inmunohistoquímicas se observó que las células expresaban neurofilamentos y sinaptofisina. El diagnóstico anatomopatológico fue de tumor neuroectodérmico primitivo.
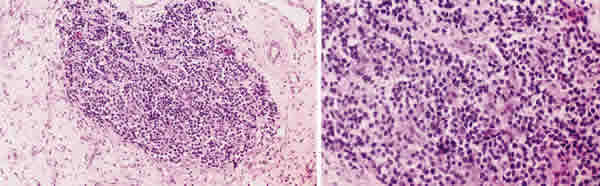
Fig. 3.Muestra de biopsia. Tinción con hematoxilina y eosina 125x (izquierda).
Tinción con hematoxilina y eosina 250x (derecha).
La evaluación sistémica (RMN craneal; TAC de tórax, abdomen, pelvis y extremidades; biopsia de médula ósea) fue negativa lo que llevó al diagnóstico definitivo de PPNET.
El paciente fue remitido al servicio de oncología donde recibió tratamiento mediante quimioterapia (QT) según el protocolo para tumores mesenquimales malignos de la Sociedad Internacional de Oncología Pediátrica (vincristina, actinomicina, fosfamida, carboplatino, epirrubicina, etopósido), entrando en remisión.
La evolución fue favorable en las múltiples revisiones, y las distintas pruebas de imagen sistémicas y orbitarias (ecografía y/o RMN) realizadas periódicamente fueron normales (fig. 4).
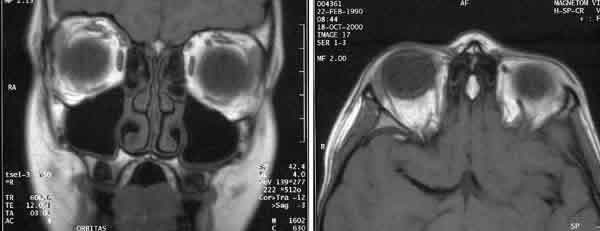
Fig. 4.RMN en T1, corte coronal (izquierda) y corte axial (derecha).
Tras permanecer siete años en remisión el paciente acude en febrero de 2004 al hospital por proptosis del OD. La RMN orbitaria mostró una masa en torno al músculo recto interno que provocaba desplazamiento lateral del contenido orbitario sin extensión ósea (fig. 5). Se realizó una nueva biopsia que tras estudio anatomopatológico se informó como recidiva de tumor neuroectodérmico ya que se observó una proliferación de células pequeñas redondeadas con abundantes figuras de mitosis y positividad para el CD19 y la sinaptofisina.
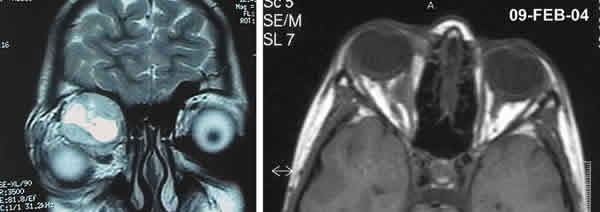
Fig. 5.RMN en T2, corte coronal (izquierda), RMN en T1, corte axial (derecha).
A pesar del tratamiento agresivo con QT (carboplatino, topotecan, ciclofosfamida), radioterapia local (dosis de 5000 cGy), y exenteración multidisciplinaria craneofacial, no se consiguió controlar el tumor apareciendo extensión cerebral y metástasis pulmonares que provocaron la muerte del paciente.
Discusión
El pico de edad de incidencia de los PPNET es en la adolescencia.
Recordemos que la localización orbitaria de este tipo de tumor es muy infrecuente, cuando se localiza en la órbita suele situarse en las paredes laterales (3).
El diagnóstico diferencial de los PPNET orbitarios debe realizarse con otros tumores de células pequeñas redondeadas incluyendo: tumor de Ewing extraóseo, metástasis de neuroblastoma, rabdomiosarcoma, linfoma, sarcoma osteogénico. Los PPNET son tumores que habían sido incluidos dentro de los tumores de Ewing extraesqueléticos con los que comparten alteraciones microscópicas, genéticas, y moleculares, distinguiéndose de éstos últimos por presentar distintos grados de diferenciación neuronal. Muchos autores consideran que los PPNET y los tumores de Ewing extraesqueléticos representan extremos del mismo grupo tumoral. Las técnicas inmunohistoquímicas y/o ultraestructurales son por tanto fundamentales para establecer el diagnóstico entre estos dos tumores y para diferenciarlos de otros tumores de células pequeñas redondeadas.
Inmunohistoquímicamente los PPNET se caracterizan por presentar positividad para: vimentina, CD19, glicoproteina p30-p32, y marcadores neuronales (neurofilamentos, sinaptofisina, cromogranina, enolasa neuronal específica). A nivel estructural en los PPNET podemos encontrar filamentos citoplasmáticos intermedios, microtúbulos citoplasmáticos y gránulos secretores.
En los PPNET orbitarios puede existir afectación ósea local y extensión extraorbitaria (1,5), pero las metástasis sistémicas son raras, solo existe un caso publicado de PPNET orbitario con metástasis hepáticas (4). Hasta la fecha, en nuestro conocimiento, tras hacer una extensa búsqueda informática, éste es el tercer caso descrito en la literatura de recurrencia orbitaria tras tratamiento (1,5) y el segundo caso descrito con metástasis sistémicas (4). De los diez pacientes publicados dos fallecieron a causa del tumor (1) pero desconocemos la evolución a día de hoy del resto de los pacientes.
Los PPNET son tumores que progresan rápidamente con mal pronóstico.
No existe consenso sobre la mejor estrategia terapéutica, algunos autores consideran la quimioterapia más menos radioterapia como primera línea de tratamiento (2).
Bibliografía
1. Howard GM. Neuroepithelioma of the orbit. Am J Ophthalmol 1965; 59: 934-937. [ Links ]
2. Singh AD, Husson M, Shields CL, De Potter P, Shields JA. Primitive neuroectodermal tumor of the orbit. Arch Ophthalmol 1994; 112: 217-221. [ Links ]
3. Kiratli H, Bilgic S, Gedikoglu G, Ruacan S, Ozmert E. Primitive neuroectodermal tumor of the orbit in an adult. A case report and literature review. Ophthalmology 1999; 106: 98-102. [ Links ]
4. Hyun CB, Lee YR, Bemiller TA. Metastatic peripheral primitive neuroectodermal tumor (PNET) masquerading as liver abscess: a case report of liver metastasis in orbital PNET. J Clin Gastroenterol 2002; 35: 93-97. [ Links ]
5. Lezrek M, Skiker H, Tachfouti S, Karim A, Karmane A, Bencherif Z, et al. Orbital primitive neuroectodermal tumor with intracanial extension. A case report. J Fr Ophtalmol 2005; 28: e8. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Ricardo Romero Martín
Urbanización Quinta del Sol, 32
28231 Las Rozas (Madrid)
España
E-mail romeromartinricardo@hotmail.com
Recibido: 15.2.06
Aceptado: 23.10.06

