










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versión impresa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.87 no.2 feb. 2012
Nuevos hallazgos tomográficos maculares en el síndrome de Alport
New macular tomography findings in Alport syndrome
Sr. Director:
El síndrome de Alport es una glomerulonefritis hereditaria progresiva caracterizada por síndrome nefrítico, sordera y alteraciones oculares. Se debe a una alteración en el colágeno tipo IV, componente fundamental de las membranas basales de todo el organismo. Las principales manifestaciones oftalmológicas descritas son retinopatía con depósitos amarillentos subretinianos -dots-flecks maculares y/o en periferia- (85% de pacientes), lenticono (25% pacientes) y menos frecuentemente distrofia corneal polimorfa posterior. La afectación ocular es más frecuente en varones y tiene una alta correlación con la afectación renal1. El desarrollo de la tomografía de coherencia óptica (OCT) ha permitido describir el adelgazamiento de la mácula temporal por disminución del grosor de las capas internas de la retina en pacientes con síndrome de Alport2,3.
Hemos analizado mediante OCT de dominio espectral 4 ojos de 2 pacientes con síndrome de Alport. El primero corresponde a un varón de 35 años con agudeza visual (AV) de 0,4 en ambos ojos. En la exploración se aprecia lenticono anterior bilateral. En el examen de fondo de ojo se observaron dots-flecks maculares temporales en anillo alrededor de la fóvea. La OCT reveló un adelgazamiento macular temporal severo dependiente de la reducción de grosor de las capas internas de la retina, concretamente nuclear interna, plexiforme interna y la membrana limitante interna (Fig. 1). El segundo caso corresponde a un varón de 36 años con AV de 0,7 en ambos ojos. En la exploración se observó lenticono anterior bilateral. La fundoscopia reveló depósitos bilaterales tipo dot-fleck en el área macular temporal. En la OCT se observó un adelgazamiento de la mitad temporal de la mácula principalmente secundaria a la disminución de grosor de la capa nuclear externa, estando el resto de capas preservadas. Tras una búsqueda exhaustiva en PubMed/Medline esta es la primera descripción de adelgazamiento macular temporal en un paciente con síndrome de Alport dependiente de capas retinianas externas (Fig. 2).
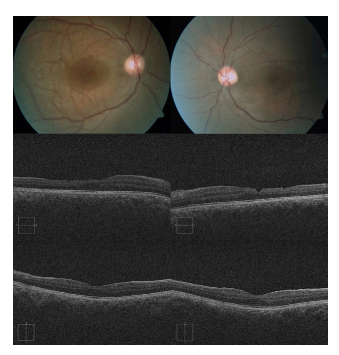
Fig. 1. Las retinografías muestran los depósitos
amarillentos subretinianos correspondientes a
dots-flecks, con una aparente hiperpigmentación
macular; la falta de foco es secundaria al acusado
lenticono. Los cortes tomográficos horizontales
centrados en la fóvea (fila central) evidencian la
asimetría secundaria a la reducción de grosor de
la retina neurosensorial temporal en ambos ojos.
En cambio, los cortes tomográficos verticales
centrados en la fóvea (fila inferior) objetivan una
retina neurosensorial de aspecto normal.
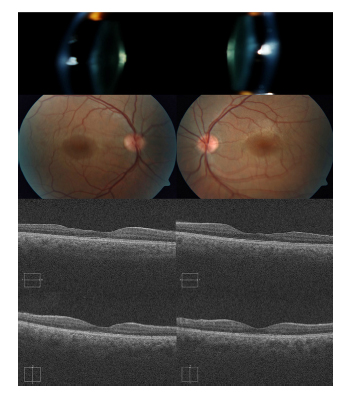
Fig. 2. En las fotografías de polo anterior se observa
el acusado lenticono causante de la falta de enfoque
de las fotografías de polo posterior. Las retinografías
muestran una hiperpigmentación macular, más acusada
por la presencia de dots-flecks parafoveales. Los cortes
tomográficos horizontales centrados en la fóvea (fila
central) evidencian la asimetría secundaria a la reducción
de grosor de la retina neurosensorial temporal en ambos
ojos. En cambio, los cortes tomográficos verticales
centrados en la fóvea (fila inferior) objetivan una
retina neurosensorial de aspecto normal.
En conclusión, la OCT de dominio espectral permite analizar en pacientes con síndrome de Alport el grado de atrofia de la retina neurosensorial cuantitativamente y evaluar las capas concretas que presentan una densidad disminuida permitiendo establecer una correlación anatómica-funcional para poder así justificar el grado de deterioro visual. Asimismo, es la primera descripción de afectación macular de predominio en capas externas asociada al síndrome de Alport.
R. Dolz-Marcoa, R. Gallego-Pinazoa, E. Francés-Muñoza, S. Martínez-Castilloa y M. Díaz-Llopisa,b
aServicio de Oftalmología, Hospital Universitario y Politécnico La Fe, Valencia, España
bFacultad de Medicina, Universidad de Valencia, España
Bibliografía
1. Hentati N, Sellami D, Makni K, Kharrat M, Hachicha J, Hammadi A, et al. Ocular findings in Alport syndrome: 32 case studies. J Fr Ophtalmol. 2008; 31:597-604. [ Links ]
2. Fawzi AA, Lee NG, Eliott D, Song J, Stewart JM. Retinal findings in patients with Alport Syndrome: expanding the clinical spectrum. Br J Ophthalmol. 2009; 93:1606-11. [ Links ]
3. Usui T, Ichibe M, Hasegawa S, Miki A, Baba E, Tanimoto N, et al. Symmetrical reduced retinal thickness in a patient with Alport syndrome. Retina. 2004; 24:977-9. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Correo electrónico: rosadolzmarco@gmail.com
(R. Dolz-Marco)

