










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkOncología (Barcelona)
versión impresa ISSN 0378-4835
Oncología (Barc.) vol.30 no.1 ene. 2007
NOTA CLÍNICA
Tumor intraabdominal desmoplásico de células pequeñas y redondas. Revisión de la literatura a propósito de un caso con estudio citológico, histopatológico, inmunohistoquímico y molecular
E. Tejerina GonzálezI; C. Corbacho CuevasI; A. López GarcíaI; C. Bellas MenéndezI; V. Sánchez TurriónII; A. Sánchez RuizIII; E. de ÁlavaIV; E. Sanz OrtegaI
IServicio de Anatomía Patológica
IIServicio de Cirugía General y Digestiva
IIIServicio de Oncología; Hospital Universitario Puerta de Hierro. Madrid
IVCIC. Universidad de Salamanca. CSIC
RESUMEN
PROPÓSITO: Revisar los hallazgos citohistopatológicos, inmunohistoquímicos y moleculares del tumor de células pequeñas redondas (DSRCT).
MATERIAL Y MÉTODOS: Varón de 18 años con masa intraabdominal epigástrica.
RESULTADOS: Tumoración constituida por una proliferación en nidos y difusa de células pequeñas indiferenciadas sobre estroma desmoplásico, con formación de rosetas, inmunorreactivas para citoqueratinas, vimentina, desmina y enolasa y la fracción WT1 de la proteína EWS/WT1. El estudio molecular demostró la traslocación t(11;22)(p13;q12).
CONCLUSIONES: La formación de rosetas y las áreas sólidas han sido descritas en otros casos de DSRCT. El producto quimérico puede detectarse mediante RT-PCR, Southern-blot, Western-blot e inmunohistoquímica. El estudio citológico está especialmente indicado en el diagnóstico de recidivas. El diagnóstico diferencial debe incluir el grupo de tumores indiferenciados de células pequeñas y redondas.
Palabras clave: Tumor intraabdominal desmoplásico de células pequeñas redondas. Inmunohistoquímica. Fusión EWT/WT1. RT-PCR.
SUMMARY
PURPOSE: To review the cyto-histopathological, immunohistochemical and molecular characteristics of small round cell desmoplastic tumor (DSRCT).
MATERIAL AND METHODS: A 18 year old male with an intraabdominal epigastric mass.
RESULTS: The tumor consisted of nests and masses of undifferentiated small round cells embedded in a desmoplastic stroma, with areas of rosette formation immuno-reactive to cytokeratins, vimentin, desmin, enolase, and WT1 fraction of the EWS/WT1 fusion protein transcript. The molecular study demonstrated the existence of the translocation t(11;22)(p13;q12).
CONCLUSIONS: The solid areas and rosette formation have been described in other cases of DSRCT. The chimeric transcription product can be detected by RT-PCR, Southern-blot, Western-blot, and immunohistochemistry. Cytology is especially useful in recurrences. The differential diagnosis should be made with the small round undifferented cell tumors group.
Key words: Intraabdominal small round cell desmoplastic tumor. Immunohiostochemistry. EWT/ WT1 fusion transcript. RT-PCR.
Introducción
El tumor desmoplásico intraabdominal de células pequeñas redondas es una neoplasia de alta agresividad descrita por Gerald y Rosai en 19891. Afecta preferentemente a varones (relación h:m de 2:1 a 10:1)2-11, en su mayoría jóvenes (media de edad, 20.8 años, con más del 80% en menores de 20 años), aunque se han descrito casos de entre 3.5 y 53 años. Hasta la fecha se han publicado unos 200 casos en la literatura2-11. Los síntomas y signos de presentación son inespecíficos e incluyen, en orden de frecuencia, dolor abdominal, pérdida de peso, hernia umbilical, ascitis, distensión abdominal, estreñimiento, hepatomegalia y esplenomegalia2-11. La mayoría se originan en la serosa de la cavidad abdominal afectando de forma difusa al peritoneo, pero se han descrito casos originados en la serosa pleural6,12 y en la túnica vaginal y región paratesticular4; existen así mismo tumores primarios no originados en serosas, incluyendo casos en fosa craneal posterior13, ovario14,15, hueso y partes blandas2, pulmón16 y glándula parótida17. Macroscópicamente son tumores grandes (hasta 18 cms de diámetro), sólidos, lobulados, blanco-grisáceos, con áreas de hemorragia y/o necrosis, en ocasiones calcificada. La neoplasia tiene una histopatología característica, estando formada por nidos bien delimitados de células pequeñas e indiferenciadas embebidas en un estroma desmoplásico densamente colagenizado. El perfil inmunohistoquímico de las células neoplásicas indica diferenciación polifenotípica, observándose coexpresión de citoqueratinas, vimentina, desmina y enolasa2-11. Esta neoplasia se asocia con la traslocación cromosómica t(11;2) (p13;q11) en la que se fusionan el exón 7 del gen EWS y el exón 8 del gen WT generando un producto quimérico de 109 pares de bases con potente actividad de factor de transcripción; dicha traslocación se considera específica de este tumor7,18-20. Se postula que el transcrito quimérico activaría el promotor del gen del factor de crecimiento-1 insulina-like, relacionado con la tumorigénesis21.
Material y métodos
El tejido tumoral fue incluido en parafina y teñido con Hematoxilina-Eosina y ácido peryódico de Schiff con/sin digestión con diastasa. El estudio inmunohistoquímico incluyó los anticuerpos reseñados en la Tabla I. Se utilizaron los sistemas Dako Envision + TM DAB System y el Vectastain ABC elite kit para la valoración inmunohistoquímica. El reordenamiento del gen EWS fue valorado con FISH mediante el uso de sondas Break-apart sobre tejido parafinado. El estudio molecular mediante RT-PCR fue realizado a partir de tejido congelado. Se utilizaron primers específicos para amplificar EWS/FLI1, EWS/ERG y EWS/WT1. La revisión de la literatura fue realizada utilizando las bases de datos MedLine y EMBASE.

Caso clínico
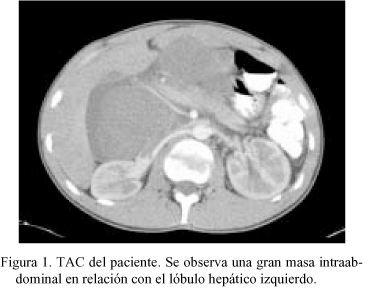
Varón de 18 años sin antecedentes personales de interés que consulta por dolor epigástrico continuo de 2-3 días de evolución, con sensación de plenitud y náuseas, sin pérdida de peso, síntomas generales ni fiebre. En la palpación profunda se aprecia una masa no móvil en el epigastrio. El TAC y la RMN (Fig. 1) muestran una tumoración polilobulada de gran tamaño localizada en el ligamento gastrohepático, que engloba la región hepatoduodenal del hilio hepático y desplaza y comprime sin infiltrar las estructuras vasculares del tronco celíaco y los órganos vecinos. No se objetiva la dependencia de un órgano en concreto aunque en algún corte parece depender del lóbulo hepático izquierdo. Las pruebas analíticas fueron normales.
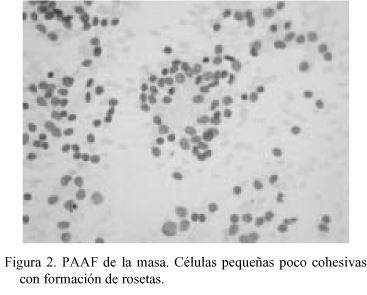
Se efectúa PAAF de la masa, tras la cual se realiza lobectomía hepática izquierda con tumorectomía y extirpación de adenopatías perihepáticas, celíacas, paracavas y retropancreáticas (8 en total), de entre 0.7 y 2.2 cm de diámetro. Tras el diagnóstico anatomopatológico el paciente recibió 17 ciclos de quimioterapia post-quirúrgica con Vincristina, Adriamicina y Ciclofosfamida alternando con Ifosfamida - Etopósido y Mesna.
Resultados
En la PAAF (Fig. 2) se obtuvieron extendidos citológicos muy celulares constituidos por células de pequeño tamaño, escasamente pleomórficas, de núcleo redondo u oval de cromatina granular con nucléolo ocasional, poco cohesivas, con ocasional formación de agregados rosetoides, inmunorreactivas para citoqueratinas. La neoplasia extirpada fue una tumoración de 22 x 16 x 10 cm multinodular, parcialmente encapsulada, constituida por tejido sólido blanco-grisáceo con áreas de necrosis y de hemorragia con degeneración quística, con buena delimitación del lóbulo hepático izquierdo, del que le separaba una pseudocápsula fibrosa. El examen microscópico (Fig. 3) mostró una proliferación neoplásica densamente celular constituida por células de pequeño tamaño con citoplasma escaso, límites celulares imprecisos y núcleos redondos u ovales de cromatina granular con ocasionales nucleólos, dispuestas en nidos sobre un estroma colagenizado en grado variable con presencia de fibroblastos activados, con áreas de necrosis. El recuento mitótico fue de hasta 19/10 cga. Además de la disposición en nidos se observaron áreas amplias de patrón difuso, con múltiples imágenes de formación de rosetas. La tinción de PAS fue negativa. El perfil inmunohistoquímico de las células neoplásicas fue (Fig. 4): AE1-AE3 positivo (focal); EMA positivo (focal); Vimentina positivo (difuso e intenso); NSE positivo (focal); Desmina positivo (difuso e intenso, con patrón en gota perinuclear); LCA, CEA , Alfa-fetoproteína, Cromogranina, Sinaptofisina, S-100, Neurofilamentos, GFAP, Mioglobina, Actina y CD99 negativos. Con estos hallazgos se emitió el diagnóstico de "tumor desmoplásico intraabdominal de células pequeñas redondas".
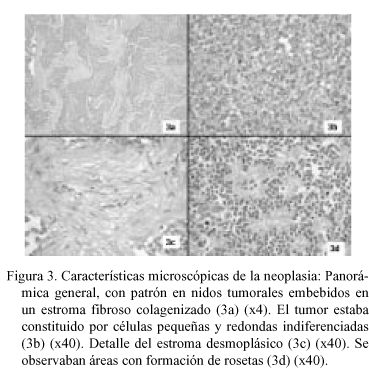
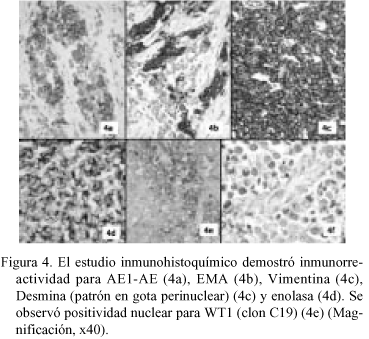
No se observó infiltración por contigüidad del tejido hepático pero sí múltiples imágenes de invasión vascular sanguínea y linfática en la cápsula hepática y en la pseudocápsula tumoral, que mostraba varias soluciones de continuidad en los nódulo neoplásicos. La totalidad de los ganglios linfáticos extirpados aparecían infiltrados por células tumorales.
Estudio molecular
Se evaluó el reordenamiento del gen EWS mediante FISH con el uso de sondas "Break-Apart", que resultó positivo. Sobre tejido congelado se realizó una RT-PCR que demostró la presencia de una banda de 268 pares de bases, concordante con el peso molecular esperado para los transcritos quiméricos derivados de la fusión EWS/WTl (Fig. 5). No se detectaron transcritos derivados de las fusiones EWS/ERG ni EWS/FLI1.

Discusión
Aunque el tumor desmoplásico de células pequeñas redondas tiene una morfología característica, en ocasiones el patrón histológico es menos típico, habiéndose descrito tumores con áreas sólidas difusas, papilares, de células claras, fusiformes, pleomórficos, rabdoides, basaloides, con formación de glándulas, rosetas y pseudorrosetas4-6,10,15,22. El perfil inmunohistoquímico de nuestro caso es superponible al descrito por otros autores; es especialmente característico el patrón en gota perinuclear de la desmina2-9,20,23. El transcrito quimérico resultante de la traslocación cromosómica t(11;12)(p13;q11) puede ser detectado mediante RT-PCR (sensibilidad: 93-96%. Especificidad: 100%) o por hibridación mediante Southern-Blot (sensibilidad: 97%)24. Los resultados negativos de la RT-PCR suelen deberse a deterioro del RNA de la muestra (por necrosis o mala preservación tisular)21. La proteína quimérica resultante de dicho transcrito puede ser detectada mediante Western blot o con técnicas inmunohistoquímicas7-9,18,19,21,23, tras el diseño de anticuerpos policlonales (C19, Santa Cruz) y monoclonales (6F-H2, Dako)23. Los anticuerpos desarrollados van dirigidos contra la región carboxiterminal (forma policlonal) o aminoterminal (forma monoclonal) de la fracción de la proteína quimérica codificada por el gen WT123. La inmunorreactividad para la fracción proteica codificada por el gen WT1 en el tumor desmoplásico de células redondas es de patrón nuclear23. La sensibilidad es de un 50% para la forma monoclonal y de entre el 88.9 y 100% para la policlonal10,21,23. Dado que el mRNA y/o la proteína codificada por el gen del tumor de Wilms están sobreexpresados en otros tumores no asociados a la traslocación descrita, es posible la inmunorreactividad para WT1 en una amplia variedad de neoplasias, con patrón tanto nuclear (mesotelioma, tumor de Wilms, carcinomas de ovario) como citoplasmática, esta última con patrón granular o difuso (carcinomas del tracto gastrointestinal, glioblastoma, osteosarcoma, sarcomas de partes blandas, melanoma, entre otros)23. Este ultimo procedimiento es de gran interés, no sólo por su fácil aplicabilidad en laboratorios de Anatomía Patológica sino también por su valor pronóstico y terapéutico, ya que WT1 está siendo evaluada como potencial diana en los tratamientos de inmunoterapia23. Aunque la práctica totalidad de los DSRCT presentan la alteración citogenética descrita (considerada diagnóstica), se han descrito formas "híbridas" en las que se solapan las características clínicas, morfológicas, inmunohistoquímicas y citogenéticas de esta neoplasia y del sarcoma de Ewing, proponiéndose para estos casos la denominación de "sarcomas bifenotípicos"22,25-27.
Los hallazgos citológicos de esta neoplasia han sido evaluados en material de punción-aspiración, derrames pleurales y ascitis28-30. El examen citológico es útil fundamentalmente en casos en los que hay dificultad en obtener biopsias y en recidivas tumorales; la necesidad de demostrar mediante inmunohistoquímica diferenciación polifenotípica hace muy difícil el diagnóstico citológico de novo30. El material de punción-aspiración es además susceptible de estudio mediante RT-PCR21.
El curso clínico es agresivo, con múltiples recidivas locales y metástasis a distancia en pulmones, hígado y ganglios linfáticos21. El tratamiento incluye cirugía y radio y/o quimioterapia21,25. El periodo libre de enfermedad es de 21-60 meses (media, 32)8. Los estudios más recientes indican que la terapia multimodal agresiva (cirugía citorreductora, poliquimioterapia y radioterapia) mejora el control tumoral24,31. La supervivencia es de 3 a 46 meses (media, 2-3 años)10,21.
El diagnóstico diferencial ha de realizarse con otros tumores de células pequeñas redondas, fundamentalmente con el sarcoma de Ewing/PNET, rabdomiosarcoma, neuroblastoma periférico "clásico", osteosarcoma de células pequeñas, condrosarcoma mesenquimal, carcinoma de células pequeñas, neuroblastoma olfatorio, carcinoma de células de Merkel, linfoma, leucemia y sarcoma granulocítico32; en algunos casos es preciso recurrir al estudio inmunohistoquímico comparativo (Tabla I). La histogénesis de estos tumores está aún por definir. La hipótesis más aceptada apunta a un origen mesotelial para estas neoplasias, considerándolo una forma de "mesotelioblastoma"; esta teoría se ve apoyada por: a) la frecuente relación con superficies serosas de estas neoplasias; b) la inmunorreactividad para WT1, presente también en el mesotelio primitivo; y 3) la coexpresión de marcadores epiteliales y mesenquimales. En contra de un origen mesenquimal iría la aparición en localizaciones sin relación con superficies serosas, la falta de inmunorreactividad para antígenos mesoteliales y la ausencia de diferenciación mesotelial en el estudio ultraestructural6,10. El estroma desmoplásico de estos tumores es resultado de la acción de la proteína quimérica derivada de la traslocación, que activaría entre otras al factor de crecimiento alfa derivado de plaquetas, con potente actividad de inducción de la proliferación fibroblástica21.
Bibliografía
1. Gerald WL, Rosai J. Case 2: Desmoplastic small round cell tumor with divergent differentiation. Pediatr Pathol 1989;9:177-183. [ Links ]
2. Adsay V, Cheng J, Athanasian E, Gerald W, Rosai J. Primary desmoplastic small round cell tumor of soft tissues and bone of the hand. Am J Surg Pathol 1999;3:1408-1413. [ Links ]
3. Amato RJ, Ellerhorst JA, Ayala AG. Intraabdominal desmoplastic small round cell tumor. Report and discussion of five cases. Arch Pathol Lab Med 2006;130:728-732. [ Links ]
4. Cummings OW, Ulbright TM, Young RH, Del Tos AP, Fletcher DC, Hull MT. Desmoplastic small round cell tumors of the paratesticular region: a report of six cases. Am J Surg Pathol 1997;21:219-225. [ Links ]
5. Dorsey BV, Benjamin LE, Rauscher F .Intra-abdominal desmoplastic small round cell tumor: expansion of the pathologic profile. Mod Pathol 1996;9:703-709. [ Links ]
6. Gerald WL, Miller HK, Battifora H, Miettinen M, Silva EG, Rosai J. Intraabdominal desmoplastic small round-cell tumor: report of 19 cases of a distinctive type of high-grade poliphenotypic malignancy affecting young individuals. Am J Surg Pathol 1991;15:499-513. [ Links ]
7. Gerald WL, Rosai J, Ladanyi M. Characterization of the genomic breakpoint and chimeric transcripts in the EWS-WT1 gene fusion of desmoplastic small round cell tumor. Proc Natl Acad Sci USA 1995;92:1028-32. [ Links ]
8. Gerald WL, Ladanyi M, de Alava E. Clinical, pathologic and molecular spectrum of tumors associated with t(11;22)(p13;q12): desmoplastic small round cell tumor and its variants. J Clin Oncol 1998;16:3028-36. [ Links ]
9. Gerald WL, Haber DA. The EWS-WT1 gene fusion in desmoplastic small round-cell tumor. Sem Cancer Biol 2005; 15:197-205. [ Links ]
10. Ordoñez NG. Desmoplastic small round cell tumor. I. A histopathologic study of 39 cases with emphasis on unusual histologic patterns. II. An ultrastructural and immunohistochemical study with emphasis on new immunohistochemical markers. Am J Surg Pathol 1998; 22:1303-13;1314-27 [ Links ]
11. Smith ME, Pelletier JP, Daniels R. Pathologic quiz case: intra-abdominal desmoplastic small round cell tumor. Arch Pathol Lab Med 2000;124:1839-1840. [ Links ]
12. Parkash V, Gerald WL, Parma A, Miettinen M, Rosai J. Desmoplastic small round cell tumor of the pleura. Am J Surg Pathol 1995; 19:659-665. [ Links ]
13. Tison V, Cerasoli S, Morigi F, Ladanyi M, Gerald WL, Rosai J. Intracranial desmoplastic small round cell tumor. Report of a case. Am J Surg Pathol 1996; 20:112-117. [ Links ]
14. Slomovitz BM, Girotra M, Aledo A. Desmoplastic small round cell tumor with primary ovarian involvement: case report and review. Gynecol Oncol 2000; 79: 124-8. [ Links ]
15. Young RH, Eichorn JH, Dickersin GR, Scully RE. Ovarian involvement by the intraabdominal desmoplastic small round cell tumor with divergent differentiation: a report of three cases. Hum Pathol 1999; 30: 430-5. [ Links ]
16. Syed S, Haque AK, Hawkins HK, Sorensen PHB, Cowan DF. Desmoplastic small round cell tumor of the lung. Arch Pathol Lab Med 2002; 126: 1226-1228. [ Links ]
17. Wolf AN, Ladanyi M, Paull G. The expanding clinical spectrum of desmoplastic small round cell tumor: a report of two cases with molecular confirmation. Hum Pathol 1999; 30:430-5. [ Links ]
18. Ladanyi M, Gerald . Fusion of the EWS and WT1 genes in the desmoplastic small round cell tumors. Cancer Res 1994; 54:2837-2840. [ Links ]
19. Sawyer JR, Tryka AF, Lewis JM. A novel reciprocal chromosome translocation t (11;22)(p13;q12)in an intraabdominal desmoplastic small round cell tumor. Am J Surg Pathol 1992;16:411-6. [ Links ]
20. Rodriguez E, Sreekantaiah C, Gerlad WL. A recurring translocation, t(11;22)(p13;q11.2) characterizes intraabdominal desmoplastic small-round cell tumors. Cancer Genet Cytogenet 1993; 69:17-21. [ Links ]
21. Lae ME, Roche PC, Jin L, Lloyd RV, Nascimento AG. Desmoplastic small round cell tumor: a clinicopathological, immunohistochemical and molecular study of 32 tumors.Am J Surg Pathol 2002; 26:823-835. [ Links ]
22. Ordi J, de Álava E, Torné A, Mellado B, Pardo-Mindán J. Intraabdominal desmoplastic small round cell tumor with EWS/ERG fusion transcript. Am J Surg Pathol 1998; 22(8):1026-1032. [ Links ]
23. Nakatsuka S, Oji Y, Horiuchi T, Kanda T, Kitagawa M et al. Immunohistochemical detection of WT1 protein in a variety of cancer cells. Mod Pathol 2006; 1-11. [ Links ]
24. Kushner, LaQuaglia M, Wollner N. Desmoplastic small round cell tumor: prolonged progression-free survival with aggressive multimodality therapy. J Clin Oncol 1996;14:1526-1531 [ Links ]
25. Katz RL, Quezado M, Senderowicz AM, Villalba L, Laskin WB, Tsokos M. An intraabdominal small round cell neoplasm with features of primitive neuroectodermal and desmoplastic round cell tumor and a EWS/FLI1 fusion transcript. Hum Pathol 1997; 28:502-9. [ Links ]
26. Rosoff PM, Hatcher S West DC. Biphenotypic sarcoma with characteristics of both a Ewing sarcoma and a desmoplastic small round cell tumor. Med Ped Oncol 2000;34:407-412. [ Links ]
27. Thorner P, Squire J, Chilton-MacNeil S. Is the EWS/FLI1 fusion transcript specific for ES and PNET? A report of four cases showing this transcript in a wider range of tumor types. Am J Pathol 1996; 148:1125-38. [ Links ]
28. Choi JK, van Hoeven K, Brooks JJ, Gupta PK. Desmoplastic small round cell tumor presenting in pleural fluids and accompanied by desmin positive mesothelial cells. Acta Cytol 1995; 39:377-378. [ Links ]
29. Crapanzano JP, Cardillo M, Lin O, Zakowski MF. Cytology of desmoplastic small round cell tumor. Cancer 2002; 96:21-31. [ Links ]
30. Dave B, Shet T, Chinoy R. Desmoplastic small round cell tumor of childhood: can cytology with immunocytochemistry serve as an alternative for tissue diagnosis? Diagn Cytopathol 2005; 32: 330-335. [ Links ]
31. Gil A, Gómez Portilla A, Brun EA, Sugarbaker PH. Clinical perspective on desmoplastic small round cell tumor. Oncology 2004; 67:231-242. [ Links ]
32. Devoe K, Weidner N. Immunohistochemistry of small round-cell tumors. Sem Diag Pathol 2000;17(3):216-224. [ Links ]
 Correspondencia:
Correspondencia:
Dra. E. Tejerina González
Servicio de Anatomía Patológica
Hospital Universitario Puerta de Hierro
C/ San Martín de Porres, 4
E-28035 Madrid
etejegon@telefonica.net
Recibido: 21.11.06
Revisado: 29.11.06
Aceptado: 11.12.06

