










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
versión impresa ISSN 1137-6627
Anales Sis San Navarra vol.35 no.2 Pamplona may./ago. 2012
https://dx.doi.org/10.4321/S1137-66272012000200002
Serie de feocromocitomas durante 15 años en Navarra
Analysis of a series pheochromocytoma cases over 15 years
J. Rojo, M. Toni, M.D. Ollero, J.J. Pineda, P. Munárriz, E. Anda
Dirección para correspondencia
RESUMEN
Fundamento. El feocromocitoma es un tumor productor de catecolaminas que procede de las células cromafines del sistema nervioso simpático. El 80-85% se localiza en la médula adrenal. El diagnóstico se confirma con la determinación de catecolaminas y metanefrinas en plasma y orina. El diagnóstico de localización debe realizarse una vez confirmado el diagnóstico bioquímico, preferentemente mediante tomografía computarizada y/o resonancia magnética. El tratamiento de elección es la cirugía vía laparoscópica.
Material y métodos. Revisión de las historias clínicas de los pacientes con diagnóstico anatomo-patológico de feocromocitoma en los servicios de Anatomía Patológica del Complejo Hospitalario de Navarra entre los años 1996 y 2010. Se realiza el análisis descriptivo de los resultados mediante el programa estadístico SPSS.
Resultados. Esta serie está formada por 43 pacientes con diagnóstico anatomo-patológico de feocromocitoma durante los últimos 15 años. La edad media de presentación es de 47 años. Entre los más jóvenes hay condensación de pacientes con determinados síndromes genéticos. El método de imagen más empleado ha sido la tomografía computarizada. Encontramos resultados contradictorios en cuanto al empleo de medicación preoperatoria. El comportamiento en todos los casos ha sido benigno.
Conclusiones. Creemos conveniente realizar estudio genético en pacientes menores de 20 años de edad. Los parámetros con mayor sensibilidad para el diagnóstico han sido normetanefrina y metanefrina en orina. El único tratamiento curativo es la cirugía, que debería realizarse preferentemente vía laparoscópica.
Palabras clave: Feocromocitoma. Catecolaminas. Metanefrinas. Médula adrenal.
ABSTRACT
Background. The pheochromocytoma is a catecholamine secreting tumour derived from chromaffin cells of the sympathetic nervous system. Eighty to eighty-five percent of these tumours are localized in the adrenal medulla. When pheocromocytomas are found outside the adrenal gland they are referred to as extra-adrenal pheochromocytomas or paragangliomas. The diagnosis is confirmed by elevation of catecholamines and the metanephrines in blood plasma and urine. Localization of the tumour should be done following biochemical diagnosis by means of CT scan and/or MRI. The treatment of choice is tumour resection by laparoscopic surgery.
Material and methods. A review was made of all patient medical histories diagnosed with pheochromocytoma confirmed by the pathology reports of Pathological anatomy of the Navarre hospital Complex (Anatomía patológica del Complejo hospitalario de Navarra A y B) between 1996 to 2010. Descriptive analysis was made using the IBM SPSS statistics program.
Results. Our series consists of 43 patients diagnosed with pheochromocytoma over a span of 15 years. The average age on presentation was 47 years. Among the younger patients specific genetic syndromes were found. Computerized tomography was the most widely used method of localization. Contradictory results were found regarding perioperative medical management protocols. All pheocromocytoma tumours in this series were benign.
Conclusions. It is advisable to carry out a genetic study on patients under twenty. The biochemical indicators with the greatest diagnostic sensitivity were the levels of normetanephrine and metanephrine in urine. Surgery was the only treatment option.
Key words: Pheochromocytoma. Catecholamines. Metanephrines. Adrenal medulla.
Introducción
El feocromocitoma es un tumor productor de catecolaminas que procede de las células cromafines del sistema nervioso simpático. El 80-85% se localizan en la médula adrenal. Los feocromocitomas de localización extraadrenal se denominan paragangliomas.
Es importante sospechar un feocromocitoma entre otras razones porque la hipertensión arterial asociada es curable, implica riesgo de muerte súbita, algunos casos son familiares y hasta un 10% pueden ser malignos1.
El diagnóstico se confirma con la determinación de catecolaminas y metanefrinas fraccionadas en plasma y orina2,3.
Una vez confirmado el diagnóstico bioquímico se realiza el diagnóstico de localización. La tomografía computarizada (TC) y la resonancia magnética (RM) deben ser utilizadas en primer lugar4,5. Cuando no es posible la localización con TC o RM, la gammagrafía con 123I-MIBG puede resultar de utilidad6,7.
El tratamiento de elección del feocromocitoma es quirúrgico, si es posible vía laparoscópica1.
Hasta una cuarta parte de los pacientes con tumores productores de catecolaminas presentan mutaciones en las líneas germinales en los genes asociados con la enfermedad. Las mutaciones identificadas con mayor frecuencia están asociadas con la enfermedad de Von Hippel-Lindau (VHL), la neoplasia endocrina múltiple tipo 2 (MEN-2), la neoplasia endocrina múltiple tipo 1 (MEN-1) y la neurofibromatosis tipo 1 (NF-1)8.
El clínico debe considerar diversos factores: localización del tumor, producción hormonal, malignidad, multicentricidad e historia familiar antes de decidir qué mutación debe estudiarse en primer lugar9.
El objetivo de este trabajo es realizar un análisis descriptivo de la serie de feocromocitomas diagnosticados en Navarra durante los últimos años y compararla con los datos publicados de otras series en la literatura al respecto. Asimismo puntualizar algunas conclusiones en cuanto al diagnóstico, tratamiento, manejo intraoperatorio y estudio genético a realizar.
Material y métodos
Se trata de un estudio retrospectivo. Para la obtención de los datos ha sido revisada la historia clínica de los pacientes con diagnóstico anatomo-patológico codificado como feocromocitoma en los servicios de Anatomía Patológica del Complejo Hospitalario de Navarra, en el periodo comprendido entre 1996 y 2010. Parte de estos pacientes habían acudido a la consulta de Endocrinología derivados desde otros servicios (Urología, Cirugía General, Cirugía Vascular y Torácica) para estudio y seguimiento.
El análisis descriptivo de los resultados se realiza mediante el programa estadístico SPSS 17.0.
Resultados
La serie del estudio está formada por un total de 43 pacientes con diagnóstico anatomo-patológico de feocromocitoma, de los cuales 32 son mujeres (74,4%) y 11 varones (25,6%). El tiempo medio de seguimiento ha sido de 6,1 años, con un rango comprendido entre 0,5 y 16 años.
Teniendo en cuenta que los datos están recogidos aproximadamente durante los últimos 15 años y que la población del área metropolitana que engloba Pamplona es de unos 300.000 habitantes, ello supone una incidencia de unos 3 casos al año (1 caso/100.000 habitantes/año).
La edad media de presentación al diagnóstico es de 47,42 +/- 18,53 (rango: 14-82) años. Por rangos de edad, puede observarse que entre los más jóvenes hay condensación de pacientes afectos de determinados síndromes genéticos que asocian feocromocitomas entre sus múltiples formas de presentación clínica.
Entre los 6 pacientes menores de 20 años de edad se encontró: 4 con MEN-2, uno con NF-1 y uno sin estudio genético, lo que supone que el 83% presentan algún síndrome genético.
Hay 13 pacientes en el rango de edad entre 20 y 40 años, entre los cuales 5 presentan MEN-2, 2 sufren NF-1 y 6 no poseen estudio genético. En este rango de edad, el 54% tienen algún síndrome genético.
El resto de pacientes de la serie presentan más de 40 años. A tres de ellos se les ha realizado estudio genético con resultado negativo. El resto no tiene estudio genético y no presentan rasgos sugestivos de ningún síndrome.
En cuanto a la etiología, 30 de los casos son, en principio, esporádicos, 12 son casos asociados (9 MEN-2 y 3 NF-1) y uno ha sido hallazgo casual en la autopsia de un paciente fallecido de ictus (Fig. 1).
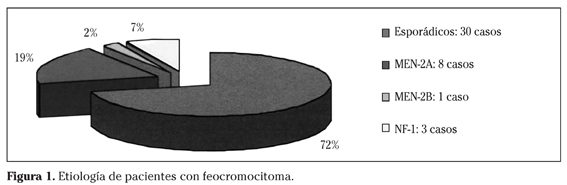
De los 43 pacientes, 37 presentan feocromocitoma unilateral (23 en la glándula suprarrenal derecha, 14 en la suprarrenal izquierda) y 6 feocromocitoma bilateral (los 6 son pacientes con MEN-2A). Por tanto, de 9 pacientes con MEN-2, el 66,6% presentan feocromocitoma bilateral.
El motivo de consulta, por orden de frecuencia, ha sido: 16 casos por crisis adrenérgica (37,2%), 12 casos como incidentaloma (27,9%), 9 casos asociado a MEN-2 (20,9%), 4 casos por dolor abdominal (9,3%), un caso por HTA mantenida (2,3%), y uno como hallazgo casual en la autopsia (2,3%) (Fig. 2).
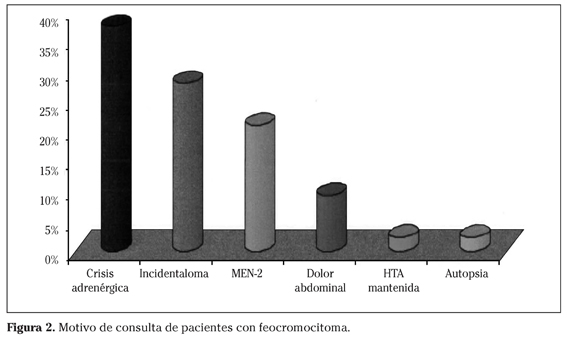
La sintomatología en el momento del diagnóstico ha sido: 77,7% HTA (37,2% HTA mantenida, 26,2% HTA paroxística, 14,3% crisis HTA sin HTA), 45,2% hiperhidrosis, 38,1% cefalea, 38,1% palpitaciones, 26,2% náuseas y/o vómitos, 23,8% nerviosismo, 23,8% mareo, 21,4% dolor precordial, 19% dolor abdominal, 14,3% pérdida de peso y 11,9% temblor.
En la tabla 1 se muestran los datos analíticos de los pacientes en el momento del diagnóstico.
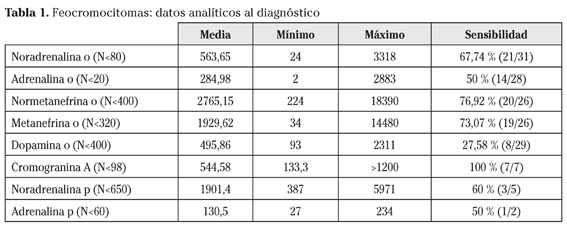
Los parámetros que han mostrado una mayor sensibilidad para el diagnóstico de feocromocitoma han sido normetanefrina y metanefrina en orina, y cromogranina A en plasma.
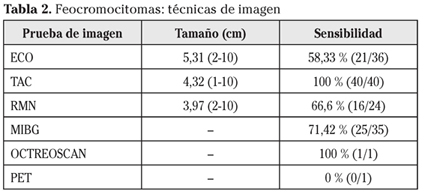
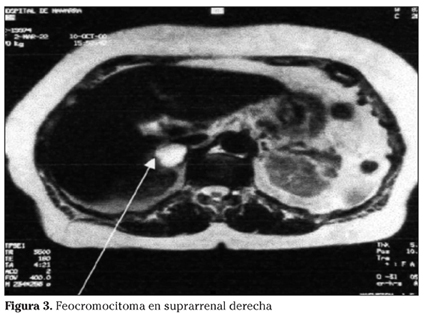
La técnica de imagen empleada más frecuentemente ha sido el TC abdominal. En la tabla 2 se muestra la correlación de tamaño entre las distintas pruebas de imagen y su sensibilidad diagnóstica (Fig. 3).
El tratamiento realizado ha sido quirúrgico en 40 pacientes: suprarrenalectomía bilateral en 5 pacientes (11,62%) y suprarrenalectomía unilateral en 35 pacientes (izquierda en 13 y derecha en 22). Una mujer embarazada ha sido intervenida mediante suprarrenalectomía unilateral derecha. La gran mayoría se ha llevado a cabo mediante cirugía abierta, sólo se han realizado 11 laparoscópicas, de las cuales, una se reconvirtió a cirugía abierta por complicaciones intraoperatorias. En la mujer embarazada se ha realizado de inicio suprarrenalectomía abierta en el primer trimestre del embarazo para evitar complicaciones para el feto. Como única prueba de imagen se había empleado la ecografía abdominal.
En un paciente se desestimó la cirugía por el riesgo quirúrgico que entrañaba su edad (82 años). Otra paciente rechazó la intervención quirúrgica. En ambos casos el tratamiento llevado a cabo fue exclusivamente médico. En otro paciente fue un hallazgo casual en la autopsia.
En 33 de los pacientes se ha realizado tratamiento farmacológico previo a la cirugía. Se ha empleado alfabloqueo solo (fenoxibenzamina) en 26 casos, con una dosis media de 28,91 (5-100) mg/24 h y una duración media de 1,2 (0,3-5) meses. En 5 pacientes se ha realizado alfa y betabloqueo (fenoxibenzamina y propranolol). Entre los pacientes normotensos se ha empleado alfabloqueo en 5 y ningún tratamiento en otros 5.
Durante la cirugía precisaron tratamiento hipotensor varios pacientes debido a oscilaciones tensionales por el manejo del tumor: nitroprusiato en 12 pacientes, fentolamina en 5 y labetalol en 8. Entre ellos, 5 eran previamente normotensos, 3 de los cuales habían recibido alfabloqueo y los otros 2 no.
A pesar del tratamiento médico previo se han documentado una serie de complicaciones quirúrgicas. En todos estos pacientes excepto en uno se había empleado tratamiento farmacológico previo: un neumotórax, dos derrames pleurales, una sección de arteria renal, dos hemorragias (uno sin tratamiento previo), tres casos de infección, dos crisis hipertensivas, cuatro casos de hipotensión, una hipoglucemia.
Desde el punto de vista anatomo-patológico, en la pieza quirúrgica de 2 casos se informa de invasión vascular. No obstante, hasta la fecha el comportamiento ha sido benigno.
En cuanto a la evolución de los pacientes con HTA y diabetes tipo 2, antes del tratamiento el 77,7% de los pacientes tenían hipertensión y tras el tratamiento esta cifra se redujo al 32,55%. Mientras que el porcentaje de diabéticos aumentó de 13,9 a 16,2% después del tratamiento.
En el momento actual se ha constatado la curación completa en 39 pacientes de la serie (90,69%). Ha recidivado únicamente un caso (2,3%), sobre la suprarrenal izquierda, que posteriormente fue reintervenido y está curado. Mientras que los 2 pacientes (4,6%) que no fueron intervenidos quirúrgicamente y que recibieron tratamiento médico combinado con fenoxibenzamina y labetalol, han fallecido presumiblemente como consecuencia directa del feocromocitoma: una paciente fallecida a los 64 años de edad consecuencia de insuficiencia cardíaca congestiva por miocardiopatía catecolamínica y la otra paciente fallecida a los 89 años por insuficiencia cardíaca descompensada por infección respiratoria.
Discusión
Esta serie difiere notablemente en la distribución de sexos respecto a otras series con un 74,4% de mujeres y un 25,6% de varones. A pesar de ello la edad media es similar a la de otras series.
Los principales motivos de consulta fueron la clínica adrenérgica (37,2%), como incidentaloma (27,9%) y asociado formando parte del MEN-2 (20,9%).
En cuanto a las cifras de tensión arterial al diagnóstico se puede diferenciar HTA mantenida (el 37% frente al 50% que describen otras series), HTA paroxística (26% frente al 33% descrito) y normotensión arterial (22% frente al 16%).
El 66,6% de los pacientes con MEN-2 presentaron feocromocitoma bilateral lo que está de acuerdo con las cifras descritas en la literatura10. Generalmente los pacientes con MEN-2 suelen desarrollar feocromocitoma posteriormente al carcinoma medular de tiroides, en nuestra serie esto había ocurrido en el 55%.
Clásicamente el diagnóstico se confirma con la determinación de catecolaminas y metanefrinas fraccionadas en plasma y orina. Las pruebas con mayor sensibilidad son las metanefrinas plasmáticas (99%), seguidas de las metanefrinas urinarias fraccionadas (97%). En cuanto a la especificidad, la mayor es la determinación de ácido vanil mandélico (95%), seguida de las metanefrinas totales urinarias (93%), las metanefrinas plasmáticas (89%) y las catecolaminas urinarias (88%). En nuestra serie, las determinaciones analíticas con mayor sensibilidad han resultado ser la cromogranina A en plasma y las catecolaminas y metanefrinas en orina. No hemos encontrado ningún falso negativo.
Dentro de las pruebas de imagen que se pueden emplear para la localización del feocromocitoma, en los algoritmos diagnósticos no está incluida la ecografía abdominal, salvo en casos en los que no pueda realizarse TC o RM. Sin embargo, ha sido la segunda técnica de imagen que más hemos empleado después del TC, porque es una prueba inocua, rápida y barata, que una vez hemos confirmado alteraciones bioquímicas nos puede servir como paso intermedio hasta la realización del TC o RM, para orientar si hay una masa a nivel suprarrenal. Su sensibilidad ha sido similar a la ofrecida por la RM. Llama la atención la baja sensibilidad de esta prueba en nuestros pacientes, comparado con la sensibilidad prácticamente del 100% que cifran otras series. El TC ha sido la prueba de localización más empleada, con una sensibilidad del 100%, similar a otras series. La gammagrafía con MIBG (yodo-131-metayodobenzilguanidina) ha presentado una sensibilidad aceptable, superior a la de la ecografía o la RM. Tan solo se ha realizado un estudio con octreoscan y un PET por lo que no podemos sacar conclusiones al respecto.
Respecto al empleo de medicación preoperatoria, al igual que en otras series, aparecen resultados contradictorios11: oscilaciones tensionales durante el manejo del tumor aparecen en pacientes con y sin tratamiento previo, y en pacientes que previamente eran normotensos e incluso habían recibido tratamiento hipotensor, mientras que crisis hipertensivas e hipotensión posterior sólo se han documentado en pacientes con tratamiento previo.
La suprarrenalectomía vía laparoscópica parece una buena opción terapéutica con escasas complicaciones, que cada vez está siendo más empleada, dejando como segunda opción la reconversión a vía abierta. Únicamente recidivó un caso (2,3% frente al 16% de otras series) en el lado izquierdo (en otras series es más frecuente en el lado derecho)12.
Los 2 pacientes (4,6%) que no fueron intervenidos quirúrgicamente y que recibieron tratamiento médico combinado con fenoxibenzamina y labetalol han fallecido como consecuencia directa del feocromocitoma.
El comportamiento en todos los casos ha sido benigno. No hemos documentado ningún caso de feocromocitoma maligno cuando en las series se describe hasta un 10%.
En nuestra serie las determinaciones analíticas con mayor sensibilidad han resultado ser la cromogranina A en plasma y las catecolaminas y metanefrinas en orina. Por tanto, deberíamos emplearlas para el despistaje de feocromocitoma en casos de alto riesgo con el fin de diagnosticar todos los casos. No obstante, en la mayoría de las ocasiones en que el diagnóstico es poco probable, la utilización de un parámetro que ofrezca mayor especificidad con aceptable sensibilidad, como la determinación de metanefrinas urinarias, puede ser de elección para evitar falsos positivos en una población de bajo riesgo13.
Respecto al empleo de medicación pre-operatoria, no está muy claro si el empleo de medicación preoperatoria consigue reducir la aparición de complicaciones. El único tratamiento curativo es la cirugía.
El porcentaje de pacientes hipertensos mejora tras el tratamiento quirúrgico. Paradójicamente aumenta el número de pacientes diabéticos (hay 6 diabéticos antes y 7 después). Debería realizarse un estudio genético en aquellos pacientes menores de 20 años, ya que hasta en el 83,3% se trata de síndromes genéticos.
Agradecimientos
Aprovechamos la ocasión para agradecer a los servicios de Anatomía Patológica del Complejo Hospitalario de Navarra su colaboración en la búsqueda de los casos.
Bibliografía
1. Ilias I, Pacak K. A clinical overview of pheochromocytomas/paragangliomas and carcinoid tumors. Nucl Med Biol 2008; 35 (Suppl. 1): S27-S34. [ Links ]
2. Pacak K, Ilias I, Adams KT, Eisenhofer G. Biochemical diagnosis, localization and management of pheochromocytoma: focus on multiple endocrine neoplasia type 2 in relation to other hereditary syndromes and sporadic forms of the tumour. Intern Med 2005: 257: 60-68. [ Links ]
3. Weise M, Merke D, Pacak K, Walther M, Eisenhofer G. Utility of plasma free metanephrines for detecting childhood pheochromocytoma. J Clin Endocrinol Metab 2002: 87: 1955-1960. [ Links ]
4. Ilias I, Pacak K. Current approaches and recommended algorithm for the diagnostic localization of pheochromocytoma. J Clin Endocrinol Metab 2004: 89: 479-491. [ Links ]
5. Mansmann G, Lau J, Balk E, Rothberg M, Miyachi Y, Bornstein S. The clinically inapparent adrenal mass: update in diagnosis and management. Endocr Rev 2004: 25: 309-340. [ Links ]
6. Jacobson A, Deng H, Lombard J, Lessig H, Black R. 123I-Meta-Iodobenzylguanidine scintigraphy for the detection of neuroblastoma and pheochromocytoma: results of a meta-analysis. J Clin Endocrinol Metab 2010; 95: 2596-2606. [ Links ]
7. Henri JL, Chen C, Carrasquillo J, Whatley M, Ling A, Havekes B et al. Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2009: 94: 4757-4767. [ Links ]
8. Casco A, Pita G, Burnichon N, Landa I, Jiménez E, Montero-Conde C et al. Genetics of pheochromocytoma and paraganglioma in spanish patients. J Clin Endocrinol Metab 2009: 94: 1701-1705. [ Links ]
9. Jiménez C, Cote G, Arnold A, Gagel R. Should patients with apparently sporadic pheochromocytomas or paragangliomas be screened for hereditary syndromes? J Clin Endocrinol Metab 2006: 91: 2851-2858. [ Links ]
10. Bravo EL, Tagle R. Pheochromocytomas: state of the art and future prospects. Endocr Rev 2003: 24: 539-553. [ Links ]
11. Kinney MA, Warner ME, Van Heerden JA, Horlocker TT, Young WF Jr, Schroeder DR et al. Perianesthesic risks and outcomes of pheochromocytoma and paraganglioma resection. Anesth Analg 2000: 91: 1118-1123. [ Links ]
12. Amar L, Servais A, Gimenez-Roqueplo AP, Zinzindohoue F, Chatellier G, Plouin PF. Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. J Clin Endocrinol Metab 2005: 90: 2110-2116. [ Links ]
13. Lenders JW, Pacak K, Walter MM, Linehan WM, Mannelli M, Friberg P et al. Biochemical diagnosis of pheochromocytoma. Which test es best? JAMA 2002: 287: 1427-1434. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Jorge Rojo Álvaro
Servicio de Endocrinología
Complejo Hospitalario de Navarra A
C/Irunlarrea, 3
31008 Pamplona
E-mail: jorgerojo23@hotmail.com
Recepción: 12 de enero de 2012
Aceptación provisional: 14 de febrero de 2012
Aceptación definitiva: 24 de febrero de 2012

