










 Servicios personalizados
Servicios personalizados
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCTION
Nucleoside and analog drugs have been widely used in cancer chemotherapy since the 1960’s for soft (leukemia and lymphoma non-Hodking) and solid (lung, liver, breast, among others) tumors treatment. These drugs exhibit several action mechanisms such as: disruption of nucleic acids synthesis, incorporation and demethylation of DNA fragments and inhibition of necessary nucleoside synthesis. Furthermore, they can present activation in cytoplasm of apoptosis factors and destabilization of mitochondrial membrane1-6.
The nucleoside anticancer drugs show several pharmacodynamic problem including low cellular incorporation across transporter channels, intracellular inactivation by nucleoside deaminase, degradation by ecto-nucleotidase enzymes, higher elimination through transporter channels and vesicles, among others. Moreover, they present very pharmacokinetic disadvantages such as low selectivity by cancer cell, higher chemical lability and biological degradation and inactivation in blood plasma3-8. Due to the considerable disadvantages of these drugs, it is recommendable that they are administered in higher doses by endovenous via; however, this administration form is responsible of the majority of many side effects (gastrointestinal disorder, neurotoxicity, nephrotoxicity and hepatotoxicity, among others) of the anticancer nucleoside3-8.
In this sense, a substantial number of studies are focused on the synthesis of nucleoside derivatives with anticancer potential, that present better physicochemical and/or biological properties, by performing lipophilic derivatives with aliphatic and aromatic acids and forming conjugates with terpenoids, heterocycles and polymers, to accomplish this, aliphatic diacids and phosphate are commonly used as linkage. With these derivatives, the purpose is to avoid the enzymatic or the chemical degradation localized in the gastrointestinal tissue, blood plasma or cytoplasm and to achieve higher cellular uptake through the cell membrane transport5-10.
Other studies focused on the nucleoside and their analogs search, show better pharmacokinetic and pharmacodynamics advantages as well as an enhancement as an antimetabolite. These studies demonstrate that the natural and synthetic derivatives of the uridine (mainly phosphorylated, sulphated and glycosylated compounds), a natural and necessary nucleoside, have cellular regulatory functions, for instance, glycosyltransferase cofactors inhibition, purinergic receptor agonists, energetic reserve (as UTP can accomplish ATP functions), DNA polymerase inhibition, among others10-13. Uridine synthetic derivatives with lipophilic substituents (C-3’-ethynyluridine, sterols, aromatic and aliphatic acids, among others) and diverse monosaccharide positions have exhibited cytotoxic activities in several tumor cell lines or better physicochemical properties with biomembrane models interaction14-18.
Furthermore, the uridine triacetate has shown modulating effects on the neurotoxicity, gastrointestinal disorders and cardiotoxicity when this is used in combinate chemotherapy with antiviral (Stavudine) and anticancer (Capecitabine, better known as fluorouracil) drugs. This uridine triacetate, known as Vistogard®, was approved by US Food and Drugs Administration (FDA) to the treatment of overdoses with these antiviral and anticancer nucleosides when they are administered for long-term19,20.
EXPERIMENTAL
Chemical
All solvents were employed at reagent grade. Uridine and cytarabine at 99% purity (Alfaesar) were employed as substrate, acetic anhydride at 99% (Merck) as agent for per-esterification reaction,4-N,N-dimethylamino-pyridine (DMAP) at 99% (Alfaesar) as nucleophilic catalyst, N-chloro-succinimide (NCS) and N-bromo-succinimide (NBS) were used for the halogenation reaction. The halogenated and/or per-acetylated derivatives of uridine and cytarabine were purified by column chromatography and thin layer chromatography using normal phase with dichloromethane/methanol and ethyl acetate/acetone mixtures as eluents of the halogenation reaction and per-acetylation, respectively. All nucleoside derivatives, halogenated and/or per-acylated, were characterized by a nuclear magnetic resonance spectroscopy on the Bruker NMR-300 and the Bruker Ascend III HD 600 MHz spectrometers, where tetramethylsilane (TMS) was used as internal standard, using deuterated dimethyl sulfoxide (DMSO-d6) and deuterated chloroform (CDCl3) as solvents. The mass spectrometry analysis was performed on the Agilent 6300-LC/MS (electrospray ionization (ESI) and Ion-Trap analyzer) and on the Agilent 6100-LC/MS (electrospray ionization (ESI) and Single Quadrupole analyzer) equipment’s, by direct injection in positive mode.
Uridine 3’,4’-acetonide (Csint)
Uridine (300 mg) was aggregated to an acetone acidulated solution with sulfuric acid4,13,17. The product (figure 1) was obtained as yellow solid (90% yield). 1H NMR (300 MHz, DMSO-d6), δ ppm (multiplicity; integration; J (Hz); position): 1.26 (s; 3H; acetonide ring), 1.48 (s; 3H; acetonide ring), 3.57 (m; 2H; H-6’ of ribose ring), 4.06 (q; 1H; J=4.2; H-5’ ribose ring), 4.74 (dd; 1H; J=6.3 y 3.5; H-4’ ribose ring), 4.89 (dd; 1H; J=6.3 y 2.6; H-3’ ribose ring), 5.63 (d; 1H; J=8.0; H-5 uracil ring), 5.82 (d; 1H; J=2.6; H-2’ ribose ring), 7.79 (d; 1H; J=8.1; H-6 uracil ring). Other NMR (13C and 2D correlations) spectroscopy characterization as was reported elsewhere17. ESI-MS (positive mode) m/z: Calculated for C12H16N2O6: 284.2652; found, 307.1 [M+Na]+.
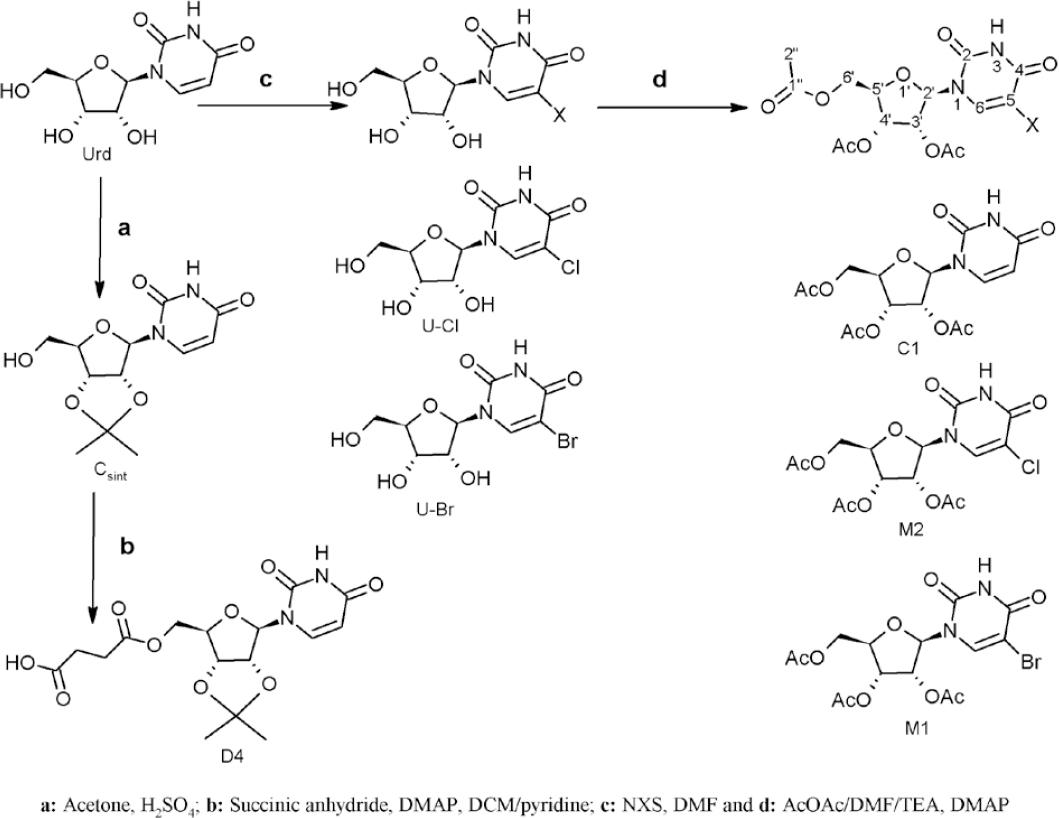
3’,4’-acetonide-6’-O-succinyl uridine (D4)
1.0 equivalents of succinic anhydride and DMAP in catalyst amount was added to uridine acetonide (300 mg) solution in DCM/pyridine mix4,9,17. The product (figure 1) was obtained as pale-yellow solid (quantitative yield). 1H NMR (300 MHz, DMSO-d6), δ ppm (multiplicity; integration; J (Hz); position): 1.26 (s; 3H; acetonide), 1.48 (s; 3H; acetonide), 2.60 (m; 4H; succinic acid), 4.40 (m; 3H; H-6’ y H-5’ ribose ring), 4.78 (m; 1H; H-4’ ribose ring), 4.93 (m; 1H; H-3’ ribose ring), 5.62 (d; 1H; J=8.0; H-5 uracil ring), 5.81 (d; 1H; J=2.6; H-2’ ribose ring), 7.80 (d; 1H; J=8.1; H-6 uracil ring). Other NMR (13C and 2D correlations) spectroscopy characterization as was reported previously17. ESI-MS (positive mode) m/z: Calculated for C16H20N2O9: 384.338; found, 407.3 [M+Na]+.
Per-acetylation reaction
The nucleoside (300 mg) was dissolved in an acetic anhydride/DMF/TEA (1:1:1) mixture, followed by the addition of DMAP (5 mol-%). The reaction mixture was stirred at room temperature between 6-8 hours 4,9,16. For chemical reaction see figures 2 and 3.
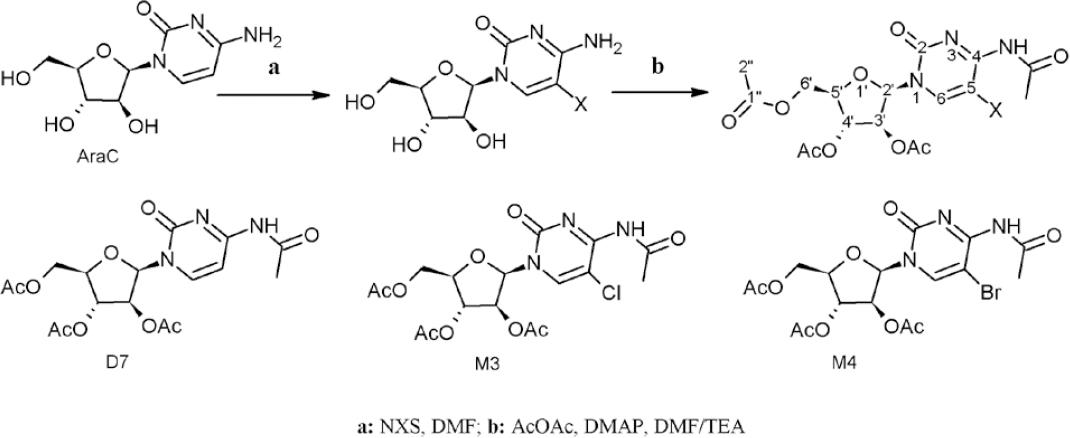
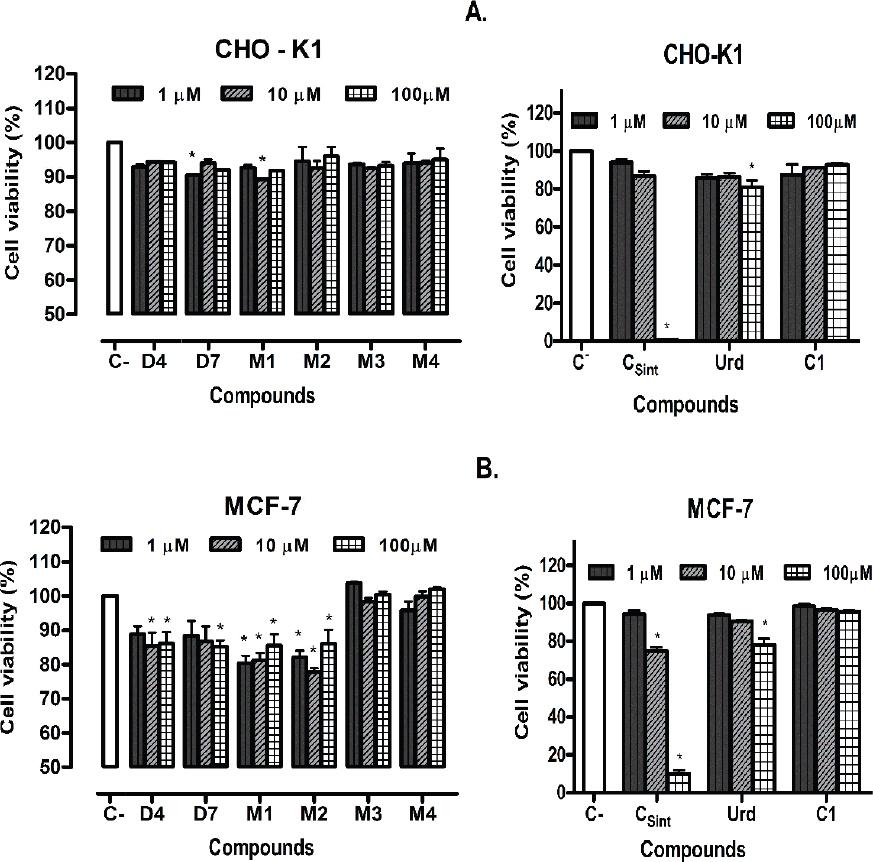
Figure 3 Cell viability of per-acetylated nucleoside (C1 and D7) an halogenated and per-acetylated derivatives (M1-M4), assessed with 1μM, 10μM and 100μM of the compounds by 48 hours. A. CHO-K1 cell line. B. MCF-7 cell line. C- (Control without treatment) and Urd: uridine. Data are shown as mean ± S.D. Statistical significance was calculated with p<0.05.
3’,4’,6’-O-triacetyl-uridine or uridine triacetate (C1)
The product was obtained as yellow solid (quantitative yield). 1H-NMR (300 MHz; DMSO-d6), δ ppm (multiplicity; integration; J (Hz); position): 2.04 (d; 6H; H-2” acetyl linked to O-3’ and O-6’ ribose ring (R.R)), 2,07 (s; 3H; position H-2” acetyl linked to O-4’ ribose ring), 4.25 (m; 3H; H-6’ and H-5’ ribose ring), 5.33 (m; 1H; H-4’ ribose ring), 5.44 (m; 1H; H-3’ ribose ring), 5.72 (d; 1H; J=8.0; H-5 uracil ring), 5.87 (d; 1H; J=5.1; H-2’ ribose ring), 7.69 (d; 1H; J=8.1; H-6 uracil ring). Other NMR (13C and 2D correlations) spectroscopy characterization as was reported elsewhere16. ESI-MS (positive mode) m/z: Calculated for C15H18N2O9: 370.3114; found, 393.1 [M+Na]+.
Halogenation reaction
The halogenation agent (NCS or NBS) was added (1.1 equivalents) on a nucleoside solution (1 mmol) in DMF, the reaction mix was stirred at room temperature for 8 hours (for chemical reaction see figures 2 and 3)21.
5-Bromo-uridine
1H-NMR (600 MHz, d6), δ ppm (multiplicity; integration; J (Hz); position): 3.57 (d, 1H; J=12.1; H-6’ ribose ring), 3.69 (d; H; J = 12.1; H-6’ ribose ring), 3.86 (d; J = 2.5; 1H; H-5’ ribose ring), 3.98 (q; J = 4.8; 1H; H-4’ ribose ring), 4.04 (q; J = 4.6; 1H; H-3’ ribose ring), 5.73 (d; J = 4.3; 1H; H-2’ ribose ring), δ 8.48 (s; 1H; H-6 uracil ring).
5-Chloro-uridine
1H-NMR (600 MHz, d6), δ ppm (multiplicity; integration; J (Hz); position): 3.58 (d; J = 10.4; 1H; H-6’ ribose ring), 3.68 (d; J = 12.0; 1H; H-6’ ribose ring), 3.86 (d; J = 2.2; 1H; H-5’ ribose ring), 3.99 (dt; J = 14.4 and 7.1 Hz; 1H; H-4’ ribose ring), 4.02 (dd; J = 9.3 and 4.5; 1H; H-3’ ribose ring), 5.73 (d; J = 4.1; 1H; H-2’ ribose ring), δ 8.40 (s; 1H; H-6 uracil ring).
5-Bromo-3’,4’,6’-O-triacetyl-uridine or 5-Bromo-uridine triacetate (M1)
The derivative was obtained coupling the halogenation and per-acetylation reactions (see reaction scheme, figure 1), this was isolated as pale-yellow solid (quantitative yield). 1H-RMN (300MHz, CDCl3), δ ppm (multiplicity; integration; J (Hz); position): 2.14 (d; 6H; H-2” (α protons) acetyl linked to O-3’ and O-6’ ribose ring), 2.18 (s; 3H; H-2” acetyl linked to O-4’ ribose ring), 4.41 (m; 3H; H-5’ and H-6’ ribose ring), 5.38 (m; 2H, H-3’ and H-4’ ribose ring), 6.12 (d; 1H; H-2’ ribose ring), 7.87 (s; 1H; H-6 uracil ring). 13C-RMN (75MHz, CDCl3), δ ppm (integration; position): 20.42 (1C; C-2” (α carbon) acetyl linked to O-6’ ribose ring), 20.52 (1C; position C-2” acetyl linked to O-3’ ribose ring), 20.94 (1C; C-2” acetyl linked to O-4’ ribose ring), 62.96 (1C; C-6’ ribose ring), 70.07 (1C; C-4’ ribose ring), 73.14 (1C; C-3’ ribose ring), 80.37 (1C; C-5’ ribose ring), 87.37(1C; C-2’ (anomeric carbon) ribose ring), 97.98 (1C; 5-C (halogenated position) uracil ring), 138.62 (1C; 6-C uracil ring), 149.70 (1C; 2-C uracil ring), 158.71 (1C; 4-C uracil ring), 169.71 (2C; C-1” acetyl linked to O-3’ and O-4’ ribose ring), 170.16 (1C; C-1” acetyl linked to O-6’ ribose ring). Calculated for C15H17BrN2O9: 449.2074; found, 472.2 [M+Na]+.
5-Chloro-3’,4’,6’-O-triacetyl-uridine or 5-Chloro-uridine triacetate (M2)
The derivative was obtained coupling the halogenation and per-acetylation reactions (see reaction scheme, figure 1), this was isolated as pale-yellow solid (quantitative yield). 1H-RMN (300MHz, CDCl3), δ ppm (multiplicity; integration; J (Hz); position): 2.16 (d; 6H; H-2” (α protons) acetyl linked to O-3’ and O-6’ ribose ring), 2.22 (s; 3H; H-2” acetyl linked to O-4’ ribose ring), 4.42 (m; 3H; H-5’ and H-6’ ribose ring), 5.38 (m; 2H; H-3’ and H-4’ ribose ring), 6.11 (d; 1H; H-2’ ribose ring), 7.78 (s; 1H; H-6 uracil ring).13C-RMN (75MHz, CDCl3), δ ppm (integration; position): 20.41 (1C; C-2” (α carbon) acetyl linked to O-6’ ribose ring), 20.51 (1C; C-2” acetyl linked to O-3’ ribose ring), 20.85 (1C; C-2” acetyl linked to O-4’ ribose ring), 62.91 (1C; C-6’ ribose ring), 70.03 (1C; C-4’ ribose ring), 73.11 (1C; C-3’ ribose ring), 80.23 (1C; C-5’ ribose ring), 87.37(1C; C-2’ (anomeric carbon) ribose ring), 110.13 (1C; 5-C (halogenated position) uracil ring), 135.99 (1C; 6-C uracil ring), 149.41 (1C; 2-C uracil ring), 158.61 (1C; 4-C uracil ring), 169.70 (2C; C-1” acetyl linked to O-3’ and O-4’ ribose ring), 170.13 (1C; C-1” acetyl linked to O-6’ ribose ring). Calculated for C15H17ClN2O9: 404.7564; found, 427.8 [M+Na]+.
4-N-acetyl-3’,4’,6’-O-triacetyl-cytarabine (D7)
The product was obtained as described in per-acetylation reaction (see scheme reaction in figure 2) and was isolated as yellow solid in quantitative yield. 1H NMR (300 MHz; CDCl3), δ ppm (multiplicity; integration; J (Hz); group; position) 1.89 (s; 3H; H-2” acetyl linked to O-3’ arabinofuranosyl ring (AF.R)), 2.09-1.98 (m; 6H; H-2” acetyl linked to O-4’ and O-6’ AF.R), 2.21 (s; 3H; H-2” acetyl linked to N-4 cytosine ring), 4.16 (t; 1H; J=10.5; H-5’ AF.R), 4.33 (t; 2H; J=12.3; H-6’ AF.R), 5.04 (d; 1H; J=10.8; H-4’ AF.R), 5.49 (d; 1H; J=2.5; H-3’ AF.R), 6.29 (d; 1H; J=3.4; H-2’ AF.R), 7.43 (d; 1H; J=7.6 Hz; H-5 cytosine ring), 7.88 (d; 1H; J=7.6 Hz; H-6 cytosine ring). 13C NMR (75 MHz, CDCl3), δ ppm (integration; position): 20.38 (1C; C-2” acetyl linked to O-3’ of AF.R), 20.60 (1C; C-2” acetyl linked to O-4’ of AF.R), 20.70 (1C; C-2” acetyl linked to O-6’ of AF.R), 24.75 (1C; C-2” acetyl linked to 4-N cytosine ring), 62.77 (1C; C-6’ AF.R), 73.97 (1C; C-3’ AF.R), 76.32 (1C; C-4’ AF.R), 81.05 (1C; C-5’ AF.R), 85.71 (1C; C-2’ AF.R), 96.45 (1C; 5-C cytosine ring), 144.77 (1C; 6-C cytosine ring), 154.72 (1C, 2-C cytosine ring), 163.25 (1C, 4-C cytosine ring), 168.26 (1C; C-1” acetyl linking to O-4’ of AF.R), 169.64 (1C; C-1” acetyl linked to O-3’ AF.R), 170.55 (1C; C-1” acetyl linked to O-6’ AF.R), 171.63 (1C; C-1” acetyl linked to 4-N cytosine ring). ESI-MS (positive mode) m/z: Calculated for C17H21N3O9: 411.3634; found, 434.1 [M+Na]+.
5-Bromo-4-N-acetyl-3’,4’,6’-O-triacetyl-cytarabine (M3)
The derivative was obtained coupling the halogenation and per-acetylation reactions (see reaction scheme, figure 2) and was isolated as yellow solid (quantitative yield). 1H NMR (300 MHz; CDCl3), δ ppm (multiplicity; integration; J (Hz); group; position) 1.88 (s; 3H; H-2” of acetyl linked to O-3’ arabinofuranosyl ring (AF.R)), 2.06-1.99 (m; 6H; H-2” acetyl linked to O-4’ and O-6’ AF.R), 2.22 (s; 3H; H-2” acetyl linked to N-4 cytosine), 4.29 (m; 3H; H-5’ and H-6’ AF.R), 5.09 (d; 1H; J=10.7; H-4’ AF.R), 5.52 (d; 1H; J=2.4; H-3’ AF.R), 6.27 (d; 1H; J=3.4; H-2’ AF.R), 8.01(s; 1H; H-6 cytosine ring). 13C NMR (75 MHz, CDCl3), δ ppm (integration; position): 20.49 (1C; C-2” acetyl linked to O-3’ AF.R), 20.72 (1C; C-2” acetyl linked to O-4’ AF.R), 20.81 (1C; C-2” acetyl linked to O-6’ AF.R), 24.88 (1C; C-2” acetyl linked to 4-N cytosine ring), 62.84 (1C; C-6’ AF.R), 73.92 (1C; C-3’ AF.R), 76.42 (1C; C-4’ AF.R), 81.12 (1C; C-5’ AF.R), 85.53 (1C; C-2’ AF.R), 96.33 (1C; 5-C cytosine ring), 144.67 (1C; 6-C cytosine ring), 155.04 (1C, 2-C cytosine ring), 163.12 (1C, 4-C cytosine ring), 168.38 (1C; C-1” acetyl linking to O-4’ of AF.R), 169.72 (1C; RO-C=O; position C-1” of acetyl linked to O-3’ of AF.R), 170.63 (1C; C-1” acetyl linked to O-6’ AF.R), 171.45 (1C; C-1” acetyl linked to N-4 cytosine ring). ESI-MS (positive mode) m/z: Calculated for C17H20BrN3O9: 490.2594; found, 513.3 [M+Na]+.
5-Chloro-4-N-acetyl-3’,4’,6’-O-triacetyl-cytarabine (M4)
The derivative was obtained coupling the halogenation and per-acetylation reactions (see reaction scheme, figure 2) and was isolated as yellow solid (quantitative yield). 1H NMR (300 MHz; CDCl3), δ ppm (multiplicity; integration; J (Hz); group; position) 1.89 (s; 3H; H-2” acetyl linked to O-3’ arabinofuranosyl ring (AF.R)), 2.07-1.98 (m; 6H; H-2” acetyl linked to O-4’ and O-6’ AF.R), 2.20 (s; 3H; H-2” of acetyl linked to 4-N AF.R), 4.27 (m; 2H; H-5’ and H-6’ AF.R), 5.06 (d; 1H; J=10.8; H-4’ AF.R), 5.49 (d; 1H; J=2.6; H-3’ AF.R), 6.27 (d; 1H; J=3.3; H-2’ AF.R), 7.96 (s; 1H; H-6 cytosine ring). 13C NMR (75 MHz, CDCl3), δ ppm (integration; position): 20.17 (1C; C-2” acetyl linked to O-3’ AF.R), 20.18 (1C; C-2” acetyl linked to O-4’ AF.R), 20.27 (1C; C-2” acetyl linked to O-6’ AF.R), 24.32 (1C; C-2” acetyl linked to N-4 cytosine ring), 62.35 (1C; C-6’ AF.R), 73.54 (1C; C-3’ AF.R), 75.97 (1C; C-4’ AF.R), 80.26 (1C; C-5’ AF.R), 84.99 (1C; C-2’ AF.R), 104.15 (1C; C-5 cytosine ring), 144.34 (1C; C-6 cytosine ring), 154.59 (1C, C-2 cytosine ring), 162.52 (1C; C-4 cytosine ring), 167.84 (1C; C-1” acetyl linking to O-4’ AF.R), 169.22 (1C; RO-C=O; C-1” acetyl linked to O-3’ AF.R), 170.12 (1C; C-1” acetyl linked to O-6’ AF.R), 171.21 (1C; C-1” acetyl linked to N-4 cytosine ring). ESI-MS (positive mode) m/z: Calculated for C17H20ClN3O9: 445.8084; found, 468,8 [M+Na]+.
Viability assay
Cell lines employed: CHO-K1 (Chinese Hamster Ovary, ATCC N° CCL61) and MCF-7 (human breast adenocarcinoma, ATCC N° HTB22). Ham F12 (Sigma) and DMEM (Sigma) were used as culture media and fetal bovine serum (FBS, Gibco, Brazil) was implemented as a supplement. Penicillin and streptomycin (Gibco) were used as antibacterial and antifungal agents, respectively. The (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide, thiazolyl blue) (MTT, Sigma). DMSO 99.9% purity (Sigma) and 2-propanol 98% purity (J. T. Baker), were employed at reagent grade. The absorbance was read in Multiskan FC Microplate Photometer–ThermoScientific spectrophotometer.
Cell culture
CHO-K1 and MCF-7 cell lines in exponential phase were cultured in Ham F-12 or DMEM medium, which were supplemented with FBS at 5% and treated with penicillin 100 U/mL and streptomycin 100 µg/mL. The cultures were incubated at 37°C for 48 hours. MCF-7 cell line was incubated in wet atmosphere with 5% CO2.
MTT assay
MCF-7 and CHO-K1 cultured cells were disseminated in 96 well plates at a cell density of 5x103 and 6x103 cells per well, respectively. The cultures were treated with concentrations of 1, 10 and 100 μM of uridine, cytarabine or their derivatives and incubated at 37°C for 48 hours, followed by an addition of 10 μL of MTT (5 mg/mL) to each plate, which were, additionally, incubated in the dark at 37ºC for 4 hours. Subsequently, 100 μL of acidified isopropanol were added to each plate and were agitated at room temperature for 1 hour. The absorbance was measured at 570 nm and cell viability percentage was calculated by three replicas of each treatment. Results were indicated with mean and standard deviation (X±SD) in at least two experiments. Statistical analysis was performed with ANOVA two-way, Bonferroni test with p<0.05 with GraphPad Prism Version 5.0 Software.
RESULTS
Chemical
The halogenation reaction with NCS and NBS allowed the selective substitution on the 5-position of uracil and cytosine ring of each nucleoside with quantitative conversions. As the esterification performed with acetic anhydride as reagent and solvent does not require heating or aggressive mediums (acidic or basic), it is an ideal reaction for highly acid labile substrates or thermo-labile, like nucleosides. This reaction is assisted with a nucleophilic catalyst, allowing the per-acylation of nucleosides, achieving the esters formation of all hydroxyl groups of ribose and arabinofuranosyl rings of uridine and cytarabine, respectively, and the amide formation on amine group of cytosine ring in the second nucleoside4,21.
The three hydroxyl groups esterification in the uridine, is simple because the OH-3’ and OH-4’ position is Z among them, and E regarding the ribose ring and other hydroxyl group (position 6’); therefore, acetyl groups are oriented in the space like scissors. Nevertheless, this reaction in the cytarabine, is very complex as the hydroxyl groups of same positions are E, and the OH-3’ is oriented Z in relation to the cytosine ring; therefore, the steric interactions are strong, even so, it was possible to obtain the per-acetylated derivative of cytarabine considering that the acetyl group is small and the anhydride acetic is used in higher amounts.
The halogenated and per-acetylated derivatives were identified comparing 1H-NMR, 13C-NMR, 1H-1H COSY, HSQC and HMBC uridine and cytarabine data (300 MHz and 600 MHz; DMSO-d6). 1H-NMR and 13C-NMR signals for the monosaccharide rings and nitrogenous bases of the nucleoside derivatives, presented a significant deviation on their chemical shift from the respective substrates (uridine and cytarabine). The NMR data and masses founded with the MS spectrometry fit with the structures proposed.
Viability assay
The results of cell viability, obtained by MTT assay with uridine and cytarabine halogenated and their per-acetylated derivatives are shown in figure 3. The per-acetylated derivatives of uridine and cytarabine, did not present significant inhibition of cell viability with the cell lines implemented (CHO-K1 and MCF-7) at the lowest concentrations evaluated (1 μM and 10 μM). Nevertheless, a higher inhibition of cell viability on both cell lines was observed for the peracetylated and halogenated nucleoside evaluated. Moreover, only the uridine acetonide exhibited a significant inhibition (higher than 80%) of cellular viabilities of CHO-K1 and MCF-7 cell lines. The uridine presented a decrease (around 20%) on MCF-7 cell viability at the highest concentration tested (100 μM).
DISCUSSION
Chemical
The per-acetylated products were obtained in quantitative yields, possibly due to the use of acetic anhydride as reagent in the acylation and in the solvent mixture, thus the effective collisions among the substrate and reagent were increased significantly. Although the amount of substrate dissolved was low, the products are highly soluble in the reaction; this property causes the displacement to the right of the equilibrium between solid and dissolved substrates. Moreover, the use of the nucleophilic catalyst (DMAP) increases the rate reaction producing a higher product formation. As the acylation reagent was in vast amount, the intermediate performed with the DMAP, is generated quickly and continuously so is available for the nucleophilic attack by alcohol or amine groups of the nucleosides4,21.
The COSY H-H spectrum for 3’,4’,6’-O-acetyl-uridine in DMSO-d6 presents at high-field, a multiplet coupling with shift δ=4.25 ppm equivalent to three protons (corresponding to overlapping of H-5’ and H-6’ protons of ribose ring) with a high deshielding signal δ=5.44 ppm (H-3’). Furthermore, this proton presents two couplings, with a shielding signal localized at δ=5.33 (H-4’) and other highly deshielded signal δ=5.87 ppm corresponding to an anomeric proton (H-2’) of the ribose ring. At down-field, a coupling between the proton appeared at δ=5.72 ppm (H-5 uracil ring) with the highest deshielding δ=7.69 (H-6 uracil ring). The COSY H-H spectrum for the 4-N-acetyl-3’,4’,6’-O-triacetyl-cytarabine in CDCl3 shows at high field that the signals for the H-5’ and H-6’protons of arabinofuranosyl ring are not overlapped, neither are the H-3’and H-4’ protons. However, it is possible to observe a coupling between protons with higher shielding δ=4.16 ppm (H-5’) of arabinofuranosyl ring and the signals at δ=4.33 ppm (H-6’). Moreover, an intense coupling is observed for the proton localized at δ=5.49 ppm (H-3’) with a high deshielding signal, δ=6.29 ppm corresponding to an anomeric proton (H-2’) of the ribose ring. At down-field, it is observed a coupling between two deshielding signals, localized at δ=7.43 ppm and δ=7.88, corresponding to H-5 and H-6 protons of cytosine ring, respectively.
In the spectrums of the halogenated and per-acetylated derivatives of nucleoside the coupling at down-field disappeared therefore, the signal for the H-6 proton is see as singlete.
The HSQC spectrum for 3’,4’,6’-O-triacetyl-uridine in DMSO-d6, presents at high-field the expected couplings between the proton signals with highest shielding of the methyl groups and the highest shielding carbons, corresponding to alpha position of the acetyl groups. Additionally, two couplings by the multiplet corresponding to H-5’ and H-6’ protons, with two kinds of carbons: a highly shielding carbon δ=62.61 ppm and another highly deshielding carbon δ=80.18 ppm, corresponding to C-6’ and C-5’ carbons of the same monosaccharide, respectively. Furthermore, it is possible to perceive the expected couplings for signals of the H-4’, H-3’ and H-2’ protons of the ribose ring with three kinds of deshielding carbons, localized at δ=70.47 ppm, δ=72.61 ppm and δ=87.17, corresponding to C-4’, C-3’ and C-2’ carbons of the monosaccharide, respectively. At down-field, it is noted a coupling among the highest deshielding proton (H-6) and a high deshielded carbon, localized at δ=139.12 ppm (C-6 carbon of uracil ring). The HSQC spectrum for the 4-N-acetyl-3’,4’,6’-O-triacetyl-cytarabine in CDCl3, shows at high field a coupling by the shielding proton at δ=4.16 ppm (H-5’) of the arabinofuranosyl ring with a high deshielding carbon δ=81.05 ppm, corresponding to C-5’ of the monosaccharide. In addition, it is possible to notice a coupling between the protons with chemical shift at δ=4.33 ppm (H-6’of arabinofuranosyl ring) and the shielding carbon at δ=62.77 ppm, corresponding to C-6’ of the monosaccharide, as well the expected couplings between the proton signals H-4’, H-3’ and H-2’ of arabinofuranosyl ring and the carbons corresponding to the same position of the monosacharide. At down-field, a coupling is detected for the proton signal of the H-5 citosine ring with deshielding carbon localized at δ=96.45 ppm, corresponding to C-5 of the same nitrogenus base and a second coupling between the proton signal with highest deshielding of the citosine ring and the highest deshielding carbon with chemical shift at δ=144.77 ppm (C-6 of nitrogenus base).
In the spectrums for the halogenated and per-acetylated compounds at down field, it is noticed only the coupling between the highest deshielding proton (H-6) of the nitrogenous base ring and a high deshielding carbon with chemical shift at δ=135-145 ppm corresponding to C-6 nitrogenous base ring.
Viability assay
The cytotoxic activity exhibited for the halogenated nucleoside and its per-acetylated derivatives has a non-significant effect. The negative result obtained is probably indicative of a phenomenon of resistance to the nucleoside for the MCF-7 cell line. This phenomenon may respond to three factors: an overexpression of receptors of tyrosine kinase (ErbB) that can accelerate the cell cycle and deactivate the cell cycle detainment performed by nucleosides22; an overproduction of the enzymatic targets pyr1-3 (the multifunctional polypeptide which comprises carbamyl-phosphate synthetase (EC 2.7.2.5), aspartate transcarbamylase (EC 2.1.3.2), dihydroorotase (EC 1.3.3.1); pyr5,6 (enzyme complex comprising orotate phosphoribosyltransferase (EC 2.4.2.10) and orotidine-5’-phosphate decarboxylase (EC 4.1.1.2), in which generally participates in the novo synthesis of pyrimidine nucleosides and in which the larger amounts of enzymes cause the inhibition of nucleoside savage route23; and when the transmembrane channels transport (of phosphoglycoprotein nature) for the nucleosides are overexpressed, as this can cause a reduction in the nucleoside cellular uptake from the exterior of the cell24.
The uridine acetonide showed a higher inhibition effect on viability of MCF-7 cell line than the uridine. This can be explained by the simple activation (phosphorylation) over OH-6’ and the hydrolysis of acetonide group, also the substituent in the acetonide derivate can provide a lipophilicity (LogP(uridine-acetonide) = 0.20, LogP(uridine) = -1.61, LogP (cytarabine) = -1.93, predicted by ACD/Labs and ChemAxon®), hence the solubility and/or interaction with the lipophilic phase of the cellular membrane can increase. In the per-acetylated and halogenated nucleoside, the adjacent chains are very small, thus the hydrolysis of the acyls groups can be viable and thermodynamically favorable, but this process may result slower than the phosphorylation in the same position3,4,8-10. Furthermore, the per-acetylated derivatives of uridine and cytarabine did not show a higher lipophilicity (LogP(uridine-triacetate) = -0.11, predicted by ACD/Labs and ChemAxon®) than the nucleosides, therefore the interaction with the lipophilic phase of membrane cell was lower than in the acetonide8-10,14. Additionally, some of the nucleoside derivatives, particularly with voluminous substituents, present a great steric impediment, which can have a inhibitory effect on the hydrolysis of 6’-acyl-ester group of the ribose, exerted by esterase enzyme and the phosphorylation over OH-6’ of the ribose ring, is essential for the activation and action mechanism of the nucleosides3,4,8,15.
CONCLUSIONS
The methods used allowed to obtain the acetylation of all hydroxyl and/or amine groups of substrates of the nucleoside type and the mono-halogenation of the nitrogenous base.
No significant effects on cell viability for the nucleoside and derivatives halogenated and per-acetylated were observed with p<0.05. Moreover, these compounds did not present a dose-response relationship, the uridine 3’,4’-acetonide showed a differential effect and a higher activity on cell viability on the MCF-7 human cancer breast cell line than on the CHO-K1 cell line. Nevertheless, the derivate 3’,4’-acetonide 6’-O-succinyl uridine showed lower activity on both cell lines than the uridine 3’,4’-acetonide. Furthermore, the breast cancer cell line MCF-7 used, according to the results, exhibits possible resistance to nucleosides and/or analogs. The per-acetylated and halogenated derivatives of the nucleosides are voluminous and present low activity, hence the decreased activation and the subsequent cytotoxic activity.

