










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkMedicina Intensiva
versión impresa ISSN 0210-5691
Med. Intensiva vol.35 no.1 ene./feb. 2011
Enfermedad de Creutzfeldt-Jakob esporádica: dos casos en medicina intensiva
Sporadic Creutzfeldt-Jakob disease: two cases in intensive care
P. Cardinal, I. Pedemonte, J. Castelli, E. Zitto, A. Cacciatore y N. Zefferino
Unidad de Cuidados Intensivos, Hospital Central de las Fuerzas Armadas, Montevideo, Uruguay
Dirección para correspondencia
RESUMEN
Las enfermedades priónicas o encefalopatías espongiformes son una familia de raras patologías neurodegenerativas caracterizadas por periodos de incubación prolongados asociados a una lenta, irreversible e invariablemente mortal evolución. En humanos se las clasifica en esporádica, adquirida y hereditaria o genética. Realizar el diagnóstico de «enfermedad de Creutzfeldt-Jakob» es un verdadero desafío para el médico intensivista dada la variabilidad en la presentación clínica y su baja incidencia. Se presentan 2 pacientes admitidos en la UCI en los que, tras descartar varias patologías, se diagnosticó con un nivel de «probabilidad», de acuerdo a la clasificación de la OMS, enfermedad de Creutzfeldt-Jakob esporádica. Se analizan aspectos diagnósticos clínicos y analíticos de la enfermedad resaltando la utilidad de la identificación de la proteína 14-3-3 en el líquido cefalorraquídeo.
Palabras clave: Encefalopatia espongiforme. Creutzfeld Jakob esporádica. Medicina intensiva.
ABSTRACT
Prion diseases or spongiform encephalopathies are a family of rare neurodegenerative diseases characterized by long incubation periods associated with slow, irreversible and invariably fatal evolution. In humans, they are classified as sporadic, acquired and hereditary or genetic. Diagnosing sporadic "Creutzfeldt-Jakob Disease" (sCJD) is a real challenge for the intensive care physician, given the variability in its clinical presentation and its low incidence. The cases of two patients admitted to the Intensive Care Unit are presented. After ruling out other diseases, they were diagnosed with sCJD with a likelihood level according to the World Health Organization Classification. Clinical and laboratory diagnostic aspects of the disease were analyzed, highlighting the utility of 14-3-3 protein identification in the cerebrospinal fluid.
Key words: Spongiform encephalopathy. Sporadic Creutzfeld Jakob. Intensive care medicine.
Introducción
Las enfermedades priónicas o encefalopatías espongiformes son una familia de raras patologías neurodegenerativas caracterizadas por periodos de incubación prolongados, asociados a una lenta, irreversible e invariablemente mortal evolución. En humanos se las clasifica en esporádica, adquirida y hereditaria o genética (Tabla 1).
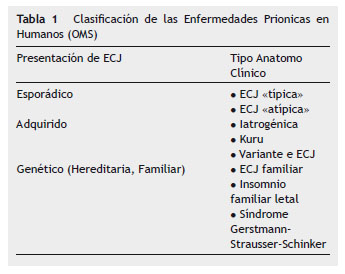
Realizar diagnóstico de enfermedad de Creutzfeldt-Jakob (ECJ), a pesar de los criterios de la OMS (Tabla 2), es un verdadero desafío para el médico intensivista, dada la variabilidad en la presentación clínica y su baja incidencia.
Se presentan dos pacientes con enfermedad de Creutzfeldt-Jakob esporádica (ECJe) admitidos a la UCI durante los años 2007 y 2008.
Caso 1: mujer de 69 años, autosuficiente para las actividades básicas de la vida diaria, sin antecedentes familiares ni personales relevantes.
Dos meses antes del ingreso hospitalario comenzó con astenia, adinamia, postración y temblor de miembros superiores. En planta de medicina, inicialmente permaneció bien orientada en tiempo y espacio aunque con episodios de confusión y desorientación. Persistió con temblor continuo de reposo en miembros superiores. Se planteó enfermedad de Parkinson y se comenzó con levodopa, sin experimentar mejoría. No presentó fiebre, leucocitosis, ni síndrome de disfunción multiorgánica. Medio interno normal. Se realizaron 2 TC (días 0 y 7) que no mostraron hallazgos de importancia.
En la evolución, de forma insidiosa, disminuyó el nivel de conciencia y presentó rigidez axial. Los movimientos anormales de miembros superiores adquirieron carácter de mioclonias.
A los 15 días del ingreso se transfiere a la UCI. El análisis citoquímico, el cultivo bacteriológico y la búsqueda de virus neurotropos por técnica de reacción en cadena de la polimerasa en líquido cefalorraquídeo (LCR) fue normal, estéril y negativo respectivamente.
Se realizó un electroencefalograma (EEG) que mostró actividad de fondo ligeramente desorganizada, con predominio de ritmos lentos de muy baja amplitud en forma bilateral. No se observaron signos de actividad epileptógena.
La RNM mostró áreas difusas e hiperintensas en la sustancia blanca subcortical, periventricular bihemisférica y cerebelosa con afectación de los núcleos caudados y lenticulares.
A los 20 días del ingreso hospitalario, en virtud del rápido deterioro neurológico, la presencia de mioclonias y los hallazgos en la RNM se planteó ECJ, en su variante esporádica dada la ausencia de antecedentes familiares y ambientales. Se realizó una nueva punción lumbar para identificación de proteína 14-3-3 que fue positiva.
Permaneció en ventilación mecánica por 20 días, se la desconecto y trasladó a planta de medicina donde falleció a los 35 días del ingreso.
Caso 2: mujer de 74 años, independiente para las actividades básicas de la vida diaria y sin antecedentes patológicos familiares ni personales a destacar. Comenzó en los dos meses previos al ingreso con astenia, adinamia, caídas frecuentes desde su altura, aumento de la base de sustentación, dismetría y temblores de predominio en miembros superiores. Consultó en emergencia, ingresando al servicio de medicina con diagnóstico probable de accidente cerebrovascular de fosa posterior. Se solicitó TC y RNM que no mostraron lesiones patológicas. Se planteó como segundo diagnóstico enfermedad de Parkinson y se comenzó con dopaminérgicos, sin experimentar ninguna mejoría.
Mala evolución, presentó alucinaciones visuales y auditivas, delirios, excitación psicomotriz y rigidez axial generalizada en ausencia de fiebre y actividad infecciosa. Se planteó como tercer diagnóstico encefalitis viral y se transfirió a la UCI. El análisis citoquímico del LCR fue normal, el cultivo bacteriológico estéril y la búsqueda de virus neurotropos negativa. Se comenzó empíricamente con aciclovir sin objetivarse mejoría.
En la UCI durante los primeros 5 días continuó disminuyendo el nivel de conciencia e incrementando el tono axial. Posteriormente, comenzó con mioclonias de miembros superiores y elementos de frontalismo (signo de Toulouse positivo y reflejo de prensión positivo).
Se realizó nueva RNM (20 días del ingreso) que mostró imágenes hiperintensas a nivel parieto-occipital derecho, asimétricas en fluid attenuated inversion recovery (FLAIR, "secuencias de inversión de fluido atenuado") (figura 1) y en diffusion weight imaging (DWI, "secuencias potenciadas en difusión") (figura 2).
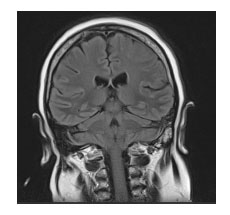
Figura 1. Imagen FLAIR que muestra una hiper
señal cortical a nivel de regiones derechas.
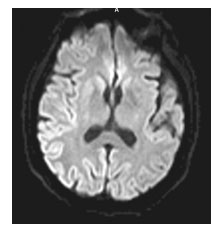
Figura 2. Mapa de coeficiente de difusión
donde se aprecia una disminución de
la intensidad de señal en las áreas derechas corticales lesionadas.
El EEG mostró actividad epileptógena generalizada intensa con descargas de complejos periódicos asociado a sufrimiento cerebral generalizado intenso.
Con la sospecha clínica de ECJe se realizó nueva punción lumbar y búsqueda de proteína 14-3-3 que fue positiva.
Permaneció en ventilación mecánica por 13 días, se la desconectó del respirador y pasó de alta al servicio de medicina donde falleció a los 45 días.
Discusión
Se presentaron dos casos de ECJe admitidos en la UCI del Hospital Central de las Fuerzas Armadas del Uruguay. El diagnóstico en ambos casos se realizó tras descartar otras etiologías y guiados por la presencia de un rápido deterioro neurológico, movimientos anormales de tipo mioclónico, imágenes de RNM y positividad de la proteína 14-3-3 en el LCR. El nivel diagnóstico fue de «probable» (Tabla 2).
Es de destacar que la presencia de movimientos anormales indujo al planteo inicial, en ambos pacientes, de enfermedad de Parkinson siendo desestimado por la rápida evolución con afectación del nivel de conciencia y la ausencia de respuesta al tratamiento instaurado.
En ambos casos se planteó posteriormente la afectación viral del encéfalo propiciando el inicio de antivirales. Se descartó por la negatividad de los estudios de reacción en cadena de la polimerasa para virus neurotropos y la refractariedad al tratamiento instaurado.
La confirmación diagnóstica de ECJe se realiza mediante el análisis histopatológico del tejido cerebral. La genotipificación del codón 129 del gen PrP (MM, MV y VV) y el análisis por western blot de la proteína priónica en el parénquima cerebral (tipo I y tipo II) permiten subtipificar a la enfermedad en seis grupos: MM1, MM2, MV1, MV2, VV1 y VV21. Las condiciones de bioseguridad requeridas para la realización del mencionado estudio así como la imposibilidad de la genotipificación del mencionado gen en nuestro medio imposibilitaron la confirmación del diagnóstico.
Recientemente, la RNM adquirió un importante rol en el diagnóstico de la ECJ a pesar de no estar incluida en los criterios de la OMS dado que presenta imágenes características desde etapas precoces de la enfermedad y permite descartar diagnósticos diferenciales2. El uso de imágenes FLAIR y/o DWI permite la detección de señales hiperintensa especialmente a nivel del núcleo caudado, del putamen y de la corteza parietal, temporal y occipital3-5. Las características observables también colaboran en la exclusión de diagnósticos diferenciales como enfermedades degenerativas, inflamatorias o infecciosas del sistema nervioso central (enfermedad de Alzheimer, demencia por cuerpos de Lewis, encefalopatía linfocítica, leucoencefalopatía multifocal progresiva, encefalitis corticoidea asociada a tiroiditis autoinmune o encefalitis de etiología desconocida)11.
En 2009 Zerr I et al1 propusieron incorporar los criterios imagenológicos al diagnóstico de ECJe. Estos incluirían la detección de señales hiperintensas en FLAIR o DWI en al menos dos regiones corticales o en el núcleo caudado y el putamen.
El EEG fue de los primeros test in vivo en ser utilizados y está incluido en los criterios diagnósticos de la OMS (Tabla 1). Los complejos trifásicos, característicos de esta enfermedad, se presentan solo en dos terceras partes de los pacientes, en algunos subtipos específicos (MM1 o MV1) y en etapas evolutivas avanzadas de la ECJe. El valor predictivo positivo puede llegar al 95%6.
En los dos casos presentados el EEG se realizó en una única oportunidad y sus hallazgos no fueron los característicos. Dadas las dificultades diagnósticas iniciales y los posteriores resultados bioquímicos del LCR e imagenológicos no se planteó la repetición del estudio.
Se han propuesto varios biomarcadores en LCR como la proteína 14-3-3, la TauS100B, la enolasa especifica de la neurona y la Tau fosforilada para el diagnóstico de la ECJe, sin embargo solo se ha incluido a la primera en los criterios diagnósticos de la OMS7. Es posible encontrarla en varios desórdenes distintos a la ECJe8 pudiendo inducir resultados falsos positivos9; sin embargo si el curso de la enfermedad es menor de un año su sensibilidad y especificidad puede llegar hasta el 92%8,10.
La ausencia de antecedentes familiares en ambos casos disminuye la probabilidad de ECJ hereditaria. En nuestra región no han sido descritos casos de ECJ adquirida por lo cual esa variante no se plantea.
La media de supervivencia de los pacientes con ECJe es de 5 meses, falleciendo el 80% durante el primer año11 tal cual sucedió en los casos presentados.
El análisis de los aspectos éticos vinculados a la limitación del esfuerzo terapéutico y al final de la vida ha sido abordado recientemente por varios grupos12,13 excediendo el propósito de la presente nota clínica. Sin embargo, destacamos que haberles brindado a ambas familias información científica actualizada, amplia disponibilidad horaria al lado del paciente e instaurar medidas de bienestar mediante un protocolo de sedación y analgesia permitió la toma de importantes decisiones de manera conjunta y no traumática entre familiares y equipo de salud.
En suma, se analizaron dos pacientes ingresados en UCI en los que la alteración de conciencia progresiva e irreversible asociada a movimientos anormales, especialmente de tipo mioclónico, y descartadas otras patologías neurológicas e infecciosas, determinaron el planteo de ECJe. El nivel diagnóstico de probabilidad se realizó mediante la identificación de proteína 14-3-3 en LCR constituyendo al momento actual el único biomarcador premórtem disponible.
Bibliografía
1. Zerr I., Kallenberg K., Summers D.M., Romero C., Taratuto A., Heneman U., et al. Update clinical diagnostic criteria for sporadic Creutzfeld-Jakob disease. Brain. 2009; 132:2659-68. [ Links ]
2. Geschwind M.D., Potter C.A., Sattavat M., García P.A., Rosen H.J., Millen B.L., et al. Correlating DWI MRI with pathologic and other features of Jakob-Creutzfeldt disease. Alzheimer Dis Assoc Disord. 2009; 23:82. [ Links ]
3. Satoh K., Shirabe S., Tsujino A., Eguchi H., Motomura M., Honda H., et al. Total tau protein in cerebrospinal fluid and diffusion-weighted MRI as an early diagnostic marker for Creutzfeldt-Jakob disease. Dement Geriatr Cogn Disord. 2007; 24:207-12. [ Links ]
4. Fujita K., Nakane S., Harada M., Izumi Y., Kaji R. Diffusion tensor imaging in patients with Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 2008; 79:1304-6. [ Links ]
5. Meissner B., Kallenberg K., Sanchez-Juan P., Collie D., Summers D.M., Almonti S., et al. MRI lesion profiles in sporadic Creutzfeldt-Jakob disease. Neurology. 2009; 72:1994-2001. [ Links ]
6. Steinhoff B.J., Zerr I., Glatting M., Schulz-Schaeffer W., Poser S., Kretzschmar H.A. Diagnostic value of periodic complexes in Creutzfeldt-Jakob disease. Ann Neurol. 2004; 56:702-8. [ Links ]
7. Zeidler M., Gibbs C.J., Meslin F. WHO Manual for Strengthening Diagnosis and Surveillance of Creutzfeldt-Jakob Disease. Geneva: WHO; 1998. [ Links ]
8. Huang N., Marie S.K., Livramento J.A., Chammas R., Nitrini R. 14-3-3 protein in the CSF of patients with rapidly progressive dementia. Neurology. 2003; 61:354-7. [ Links ]
9. Chapman T., McKeel D.W., Morris J.C. Misleading results with the 14-3-3 assay for the diagnosis of Creutzfeldt-Jakob disease. Neurology. 2000; 55:1396-7. [ Links ]
10. Van Everbroeck B., Michoette A., Sciot R., Godfraind C., Deprez M., Quoilin S., et al. Increased incidence of esporadic C-J disease in the age groups between 70 and 90 years in Belgium. Eur J Epidemiol. 2006; 21:443-7. [ Links ]
11. Nuñez Cuerda L., Matías Salce M.L., Colas Rubio J., Martín Barranco M.J., Marcos Sánchez F. Enfermedad de Creutzfeldt-Jakob: dos nuevos casos en Talavera de la Reina. Rev Clin Esp. 2008; 208:193-6. [ Links ]
12. Monzón Marín J.L., Saralegui Reta I., Abizanda Campos R., Cabré Pericas L., Iribarren Diarasarri S., Martín Delgado M.C., et al. Recomendaciones de tratamiento al final de la vida del paciente crítico. Med Intensiva. 2008; 32:121-33. [ Links ]
13. Santos D., Della Valle A., Barlocco B., Pereyra J., Bonilla D. Sedación paliativa: experiencia en una unidad de cuidados paliativos de Montevideo. Rev Med Urug. 2009; 25:78-83. [ Links ]
 Dirección para correspondencia:
Dirección para correspondencia:
pablocardinal@hotmail.com
(P. Cardinal)
Recibido 27 Enero 2010
Aceptado 16 Febrero 2010

