










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNefrología (Madrid)
versión On-line ISSN 1989-2284versión impresa ISSN 0211-6995
Nefrología (Madr.) vol.34 no.2 Cantabria 2014
https://dx.doi.org/10.3265/Nefrologia.pre2014.Feb.12157
CARTAS AL DIRECTOR - CASOS CLÍNICOS BREVES
Afectación multigénica en el síndrome nefrótico congénito
Multigene involvement in congenital nephrotic syndrome
Dirección para correspondencia
Sr. Director:
El síndrome nefrótico congénito (SNC) es una enfermedad grave y rara de herencia autosómica recesiva y monogénica en la mayoría de las ocasiones. Existen diferentes genes implicados, siendo los más frecuentes NPHS1, NPHS2, WT1 y LAMB21,2. Clínicamente se manifiesta con proteinuria masiva, edemas generalizados, hipoalbuminemia e hipertrigliceridemia de aparición en los primeros tres meses de vida3. Presentamos el primer caso de un paciente afecto de SNC con alteración multigénica de tres de los cuatro genes más frecuentes.
Caso clínico
Se trata de un varón de un mes de vida, de origen marroquí, que acude a Urgencias por presentar vómitos, rechazo del alimento y distensión abdominal de 4 días de evolución. El embarazo fue controlado, sin existir antecedentes obstétricos ni perinatales de interés, naciendo a término y con un peso adecuado a su edad gestacional. En la exploración destacaba un regular estado general, con palidez cutáneo-mucosa y edemas generalizados de predominio en las extremidades inferiores. Se auscultó un soplo sistólico grado IV/VI polifocal. El abdomen estaba distendido, con presencia de red venosa superficial y ascitis. El examen de laboratorio mostró la presencia de anemia normocítica normocrómica, leucocitosis con fórmula normal, creatinina menor de 0,2mg/dl y urea de 10mg/dl, aumento del colesterol y los triglicéridos, y disminución de proteínas totales y albúmina, además de hiponatremia, hipopotasemia e hipocalcemia (tabla 1). La hormona paratiroidea se encontraba levemente elevada. En orina presentaba una proteinuria en rango nefrótico con índice proteinuria/creatinuria de 33,7.
Tabla 1. Parámetros analíticos al ingreso. 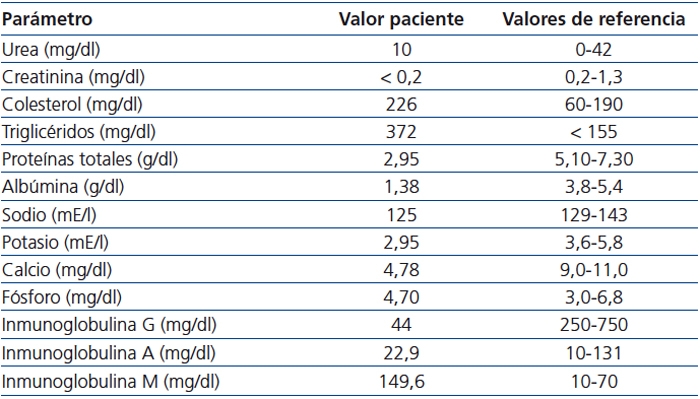
Ante la sospecha de SNC, se inició terapia intensiva diurética y antiproteinúrica, profilaxis antitrombótica, tratamiento adyuvante con alfacalcidiol, hierro, carbonato cálcico y levotiroxina, además de nutrición enteral con fórmula hiperproteica e hipercalórica. Precisó tratamiento con seroalbúmina y eritropoyetina. En el estudio cardiológico se diagnosticó estenosis pulmonar valvular moderada y comunicación interauricular. En el estudio genético se encontraron las mutaciones tipo Frameshift para el gen NPHS1, tipo Intronic Variant para el gen NPHS2 y tipo Missense para el gen WT1.
A los tres meses de edad reingresó por presentar estatus convulsivo secundario a hipocalcemia grave. Debido a los aportes elevados de calcio intravenoso que precisó para su control a través de un acceso venoso periférico, se produjo una quemadura de tercer grado que necesitó la colocación de injerto cutáneo.
Precisó cuidados intensivos en dos ocasiones a los 4 y 6 meses de edad por sepsis secundaria a Staphylococcus hominis y Enterococcus faecalis, respectivamente, que se resolvieron con antibioterapia empírica y posteriormente según antibiograma.
Finalmente, el paciente falleció a los 8 meses de edad debido a una neumonía bilateral asociada a neumotórax con descompensación de su patología de base que provocó una hipoxemia refractaria.
Discusión
El SNC puede sospecharse en el período prenatal por niveles elevados de alfafetoproteína, manteniendo cifras normales de colinesterasa a partir de la semana 15 de gestación en líquido amniótico y sangre materna2,4. En el nacimiento orienta la asociación de prematuridad y placenta grande5, datos no presentes en nuestro paciente.
La causa más frecuente de esta entidad es la mutación del gen NPHS1, de herencia autosómica recesiva, y responsable de la codificación de la nefrina2,5,6. Esta alteración es particularmente común en Finlandia, por lo que se le ha dado el nombre de SNC de tipo finlandés. El gen NPHS2 codifica la proteína podocina y es la causa más común de corticorresistencia en la infancia7. Por otra parte, el gen WT1 juega un papel crucial en el desarrollo embrionario del riñón y los genitales, y se ha relacionado con la presentación de síndromes tales como el WAGR, Denys-Drash o Frasier1,8. Se considera una enfermedad monogénica y no se ha descrito en la literatura ningún caso con afectación de más de dos genes implicados, por lo que se desconoce la relevancia clínico-patológica del hallazgo encontrado en nuestro caso.
El método de elección para su diagnóstico es el análisis genético, aunque previamente han de descartarse las causas secundarias, como infecciones congénitas, enfermedades autoinmunes o exposición a tóxicos durante el embarazo9. Su conocimiento puede ser útil en el manejo, el pronóstico y el seguimiento del paciente, y clave para ofrecer consejo genético a la familia10. La biopsia renal no revela la etiología del SNC, ya que los hallazgos histológicos pueden aparecer en distintas entidades2. El SNC no suele asociarse a malformaciones cardíacas, exceptuando una frecuencia elevada de hipertrofia ventricular11. Nuestro paciente presentaba estenosis valvular pulmonar y comunicación interauricular, defectos poco descritos en la literatura.
Las complicaciones más frecuentes son las infecciones bacterianas, sobre todo por microorganismos gramnegativos2. Nuestro caso presentó dos cuadros sépticos por bacterias grampositivas, hallazgos menos frecuentes, pero comunicados en otras publicaciones2.
El número de hospitalizaciones en estos pacientes sigue siendo muy elevado debido a su difícil manejo terapéutico y a las complicaciones, lo cual supone un detrimento en la calidad de vida12. Es posible que una afectación multigénica pueda implicar una mayor gravedad clínica, pero al no existir datos descritos similares al nuestro no podemos afirmar estas consideraciones. El objetivo del tratamiento es controlar el edema, prevenir y tratar las complicaciones, y proporcionar una nutrición adecuada para maximizar el crecimiento y permitir demorar el tratamiento sustitutivo, aunque, en la mayoría de los casos, el trasplante renal es la única medida terapéutica curativa3,5,6.
En conclusión, el SNC es una patología grave, rara y de difícil manejo, en cuya etiología pueden verse involucrados varios genes, que quizá tenga implicaciones relevantes en la gravedad clínica y complicaciones asociadas.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
Elena Cobos-Carrascosa, Ana Campos-Aguilera, Antonio Daza-Torres
Sección de Nefrología Pediátrica. Hospital Torrecárdenas. Almería
Referencias Bibliográficas
1. Fencl F, Malina M, Stará V, Zieg J, Mixová D, Seeman T, et al. Discordant expression of a new WT1 gene mutation in a family with monozygotic twins presenting with congenital nephrotic syndrome. Eur J Pediatr 2012;171:121-4. [ Links ]
2. Mehrazma M, Otukesh H, Madani A, Hooman N, Bedayat A, Dianati Maleki N, et al. Histopathologic and clinical findings of congenital nephrotic syndrome in iranian children: A study of two centers. Iran J Kidney Dis 2012;6:426-31. [ Links ]
3. Canalejo González D, González Rodríguez J, Navas López V, Sánchez Moreno A, Fijo López-Viota J, Martín Govantes J. Evaluación de las estrategias terapéuticas en el síndrome nefrótico congénito tipo finlandés. An Pediatr (Barc) 2006;65:561-8. [ Links ]
4. Gigante M, Greco P, Defazio V, Lucci M, Margaglione M, Gesualdo L, et al. Congenital nephrotic syndrome of Finnish type: Detection of new nephrin mutations and prenatal diagnosis in an italian family. Prenat Diagn 2005;25:407-10. [ Links ]
5. Benoit G, Machuca E, Heidet L, Antignac C. Hereditary kidney diseases: Highlighting the importance of classical mendelian phenotypes. Ann N Y Acad Sci 2010;1214:83-98. [ Links ]
6. Badoe E, Kumoji R. Congenital nephrotic syndrome of the finnish type. Ghana Med J 2008;42:42-4. [ Links ]
7. Dámaso EO, González NS, Pérez JCR. Síndromes nefróticos hereditarios. Podocitopatías. Nefrologia 2011;2(1):21-8. [ Links ]
8. Hinkes BG, Mucha B, Vlangos CN, Gbadegesin R, Liu J, Hasselbacher K, et al. Nephrotic syndrome in the first year of life: Two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics 2007;119:e907-19. [ Links ]
9. Kupferman JC, Spitzer ED, Stokes MB. A critically ill infant with sepsis, respiratory failure, and anasarca. Am J Kidney Dis 2013;61:22-5. [ Links ]
10. Kaukinen A, Kuusniemi AM, Lautenschlager I, Jalanko H. Glomerular endothelium in kidneys with congenital nephrotic syndrome of the Finnish type (NPHS1). Nephrol Dial Transplant 2008;23:1224-32. [ Links ]
11. Grech V, Chan MK, Vella C, Attard Montalto S, Rees P, Trompeter RS. Cardiac malformations associated with the congenital nephrotic syndrome. Pediatr Nephrol 2000;14:1115-7. [ Links ]
12. Finn LS, Symons JM, Smith JM. Nephrotic syndrome in the newborn. Am J Kidney Dis 2003;42:1318-23. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Elena Cobos-Carrascosa,
SECCIÓN DE NEFROLOGÍA,
Hospital Torrecárdenas,
Paraje Torrecárdenas, s/n,
04009, 04721, Almería
E-mail: krass10@hotmail.com

