










 Servicios personalizados
Servicios personalizados
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCTION
Phenylketonuria (PKU) is characterized as an innate, autosomal recessive alteration of phenylalanine metabolism caused by variants in the gene 12q22-q24.2 that encodes phenylalanine hydroxylase (PAH) (1). PAH deficiency leads to accumulation of the amino acid phenylalanine in the blood and, consequently, in the cerebrospinal fluid, as it is not metabolized to tyrosine (2,3). In PKU, tyrosine becomes an indispensable amino acid as it is not provided endogenously by hydroxylation of phenylalanine, or such conversion occurs to a very limited degree (4,5).
Early diagnosis and proper treatment prevent major neurocognitive deficits (5,6). Treatment focuses on a natural low-protein diet throughout life, with the goal of reducing phenylalanine intake (6). And, despite the fact that guidelines on the dietary treatment of PKU (2,3,7-10) are similar worldwide, the dietary intake of these individuals can vary widely because of different cultural eating habits, lifestyle, access to specialized health services with multidisciplinary team, and metabolic formulas available in each location (11,12).
Thus, knowing the food intake of this population makes it possible to adjust the nutritional treatment and create guidelines and strategies for food and nutrition education to have an impact on the overall health of individuals with PKU. Thus, the objective of this study was to analyze the evidence on food intake of individuals with PKU.
MATERIAL AND METHODS
An integrative literature review was conducted based on the analysis of articles that assessed the dietary intake of individuals with PKU with the following guiding question: “What is the nutritional composition of the diet of individuals with PKU?” The development of this study followed the steps proposed for the development of an integrative review (13). Initially, an identification of the topic and a formulation of the research question were performed, followed by the establishment of eligibility criteria; database search; data analysis and interpretation; presentation, interpretation and discussion of results.
The study included original articles, available online and in full, in any language, with no time limit, that addressed the energy and macronutrient intake of children, adolescents and/or adults with PKU without the use of drugs (e.g., sapropterin hydrochloride, pegvliase-pqpz). Articles whose content did not address the guiding question, studies with pregnant women, infants or animals, consensus statements, guidelines, theses, dissertations, literature reviews, case studies, and abstracts were excluded. If the study was an intervention study, only the baseline data of PKU patients were evaluated.
The step corresponding to the search for studies was performed in March and April 2022, in the Pubmed, Bireme and Science Direct databases, using the descriptors: “macronutrients”, “eating”, “nutritional status and “diet”. These descriptors were associated with the term “Phenylketonuria” with the help of the Boolean operators “AND” and “OR” (Table I).

The results were imported into the Excel® program and, after checking for duplicates, they were summarized in another Excel® spreadsheet. The analysis of the studies was performed by two independent evaluators, starting with the analysis of the title and then the abstract, using a standardized eligibility form, according to the model proposed by the Brazilian Ministry of Health (14). For this analysis, the following evaluation criteria were established: type of study and food consumption in PKU.
Next, the selected studies were compared between the two researchers to verify the agreement of the studies included in this review. If there were differences, they were discussed based on the pre-established inclusion criteria described above. After this step, the articles were read in their entirety. The methodological approach, data extraction and writing of the findings followed the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines (15). Data were organized and summarized in tables with information about the studies, (author’s name and year of publication), sample, method of food intake assessment, and main results found (energy intake, protein, glucose, lipid, fiber, blood levels-metabolic control, and dietary intake of phenylalanine. From the results found, the studies were categorized into three tables.
RESULTS
The initial search resulted in 384 articles, which after exclusion of duplicate documents totaled 365 studies. With the application of the eligibility criteria, 63 articles were selected for analysis. However, after full reading 36 documents were excluded, resulting in a final analysis of 27 articles (Fig. 1).
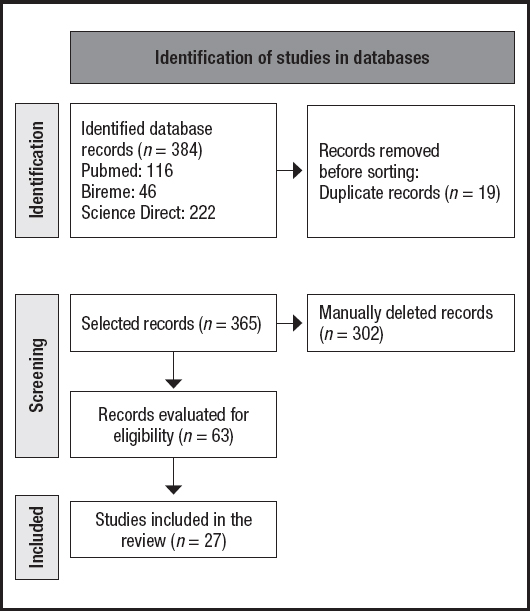
Table II summarizes the studies found and published in the last 30 years, with more intensity in the last 10 years. The sample size of the studies ranged from 10 to 101 individuals with PKU, aged between 1 and 52 years. The countries with the highest concentration of studies were the United States (n = 5), followed by Brazil (n = 3) along with Italy (n = 3) and the United Kingdom (n = 3).
Table II. Characterization of the selected studies.
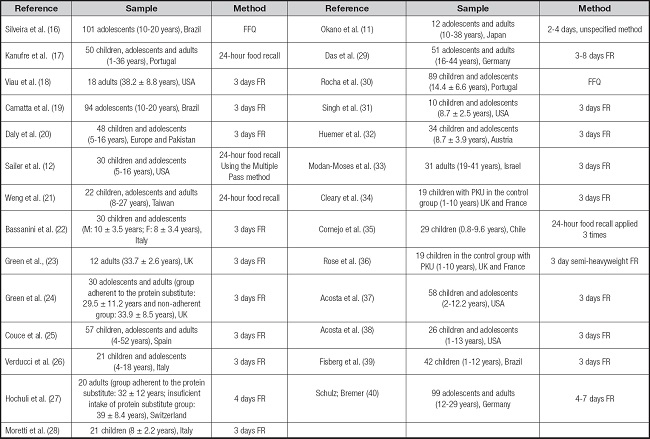
PKU: phenylktonuria; FR: food record; FFQ: food consumption frequency questionnaire.
In most studies, the instrument to assess food intake was the three-day food record. Only one study used the 24-hour food recall method plus the Multiple Pass method and few used the food intake frequency questionnaire (Table II).
The energy intake of individuals with PKU cited in the studies ranged from 1160 to 2700 kcal/day (Table III). Among the studies that evaluated and compared the energy intake of individuals with PKU with Recommended Dietary Intake (RDA) values, seven studies found insufficient energy intake (18,31,34,37-40) and one observed adequate intake (11). When comparing different groups of individuals with PKU or with individuals with hyperphenylalanine, most (n = 6) articles observed no statistically significant difference (p > 0.05) in energy intake between the groups (16,24,26,36-38) and two studies found insufficient energy intake (29,33), especially in individuals not adherent to nutritional therapy for PKU (33). Also, three studies found no statistically significant difference (p>.05) between the energy intake of individuals with PKU compared to the healthy control group (21,28,35), and only one study found lower consumption (12). The other studies did not compare the energy values found since it was not the objective of their study.
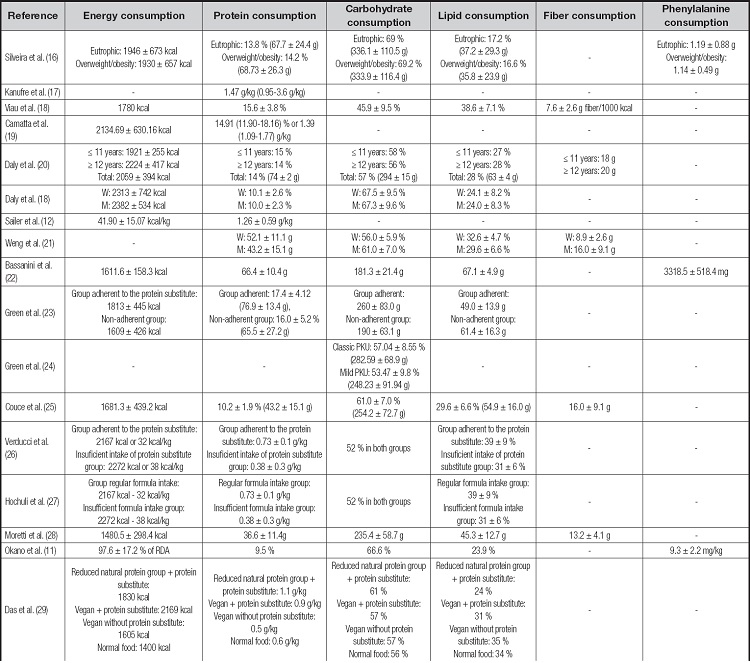
Table III (cont). Analysis of dietary intake of different individuals with phenylketonuria.
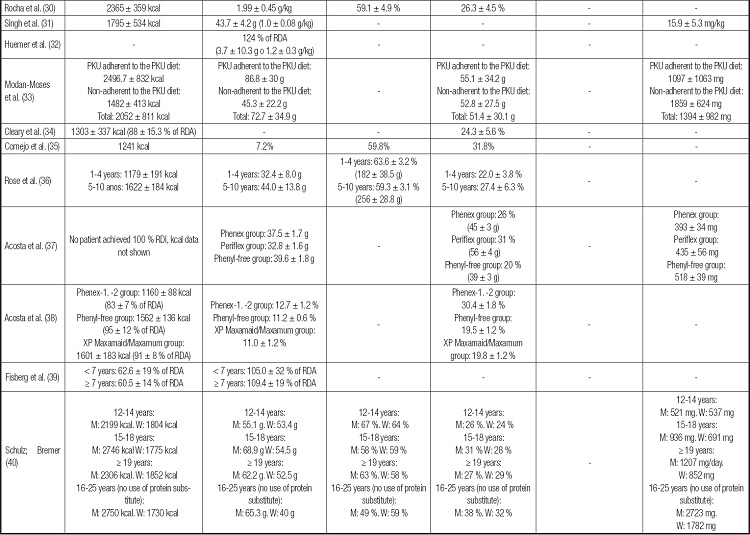
-: not assessed or not present; PKU: phenylktonuria; TEV: total energy value; BMR: basal metabolic rate; RDA: Recommended Dietary Allowances. RDI: Reference Daily Intake; M: men; W: women.
The protein intake of individuals with PKU ranged from 32.4 g to 76.9 g/day in the studies found (Table III). Among the studies that compared the protein values found with the reference values ((RDA)/guidelines for PKU), seven studies found adequate protein intake (17,18,31,32,37,39,40) and two verified insufficient intake (27,29). Studies that compared protein intake with a healthy control group or hyperphenylalanine group (n = 5) showed lower intake in the PKU group (12,26,28,35), and only one study (21) showed similar intakes. Studies that compared protein intake between different groups of individuals with PKU showed similar protein intake (16,24,36,38), except Modan-Moses et al. (33), who found higher protein intake in the group of individuals adherent to nutritional treatment for PKU. The other studies did not compare the values found.
The carbohydrate intake reported in the studies was between 45.9 % and 69.2 % of the total energy value (TSV) of individuals with PKU (Table III). Among the studies that evaluated different groups of individuals with PKU, there was evidence of higher carbohydrate intake in patients adherent to the recommended dietary treatment (24,29) and in the younger groups (ages between 12 and 14 years) (40) but other studies found no statistically significant difference between different groups with PKU over 5 years (16,36). Some works also showed higher carbohydrate intake in individuals with PKU compared to healthy controls (12,25,26,28,30) or compared to Dietary Reference Intakes (DRI) (11,16). Few studies (n = 3) found adequate glycemic intake in the phenylketonuric population (18,27,35).
Lipid intake of individuals with PKU ranged from 16.6 % to 39 % (Table III). Studies with different groups of individuals with PKU found no statistically significant difference in lipid intake (33,34,36,38). However, it appears that PKU patients adherent to dietary treatment had lower lipid intake than non-adherent PKU patients (24,27,29). A lower lipid intake was observed in individuals with PKU compared to healthy control groups (12,28) and compared to reference dietary recommendations (16,40), while other authors found no differences with respect to reference dietary recommendations (11,26,30,37).
It is important to mention that Cornejo et al. (35) found no statistically significant difference in the percentage of total fats consumed between the PKU group and the healthy control group; however, they found differences between the types of fats consumed, with higher consumption of polyunsaturated fats and linoleic acid and lower consumption of saturated fats, monounsaturated fats and alphalinoleic acid in the PKU group. Only one study demonstrated higher fat intake in individuals with PKU compared to the reference dietary recommendations (18).
The fiber intake by individuals with PKU ranged from 7.6 g to 20 g/day (Table III). The results showed insufficient consumption in relation to dietary recommendations (18), adequate in others (20,26), and higher than the healthy group evaluated (28), which shows the need for further studies in this area.
Phenylalanine intake was assessed by few studies (n = 7) and showed intake between 393 and 2723 mg/day (Table IV). The studies that compared different groups with PKU (n = 3) found no statistically significant difference in phenylalanine intake (16,33,37). However, the studies that compared the values of phenylalanine consumed with the reference recommendations, mostly verified adequate consumption (11,31,40), while only one study found high consumption (23).
Table IV. Analysis of the metabolic control of individuals with phenylketonuria.
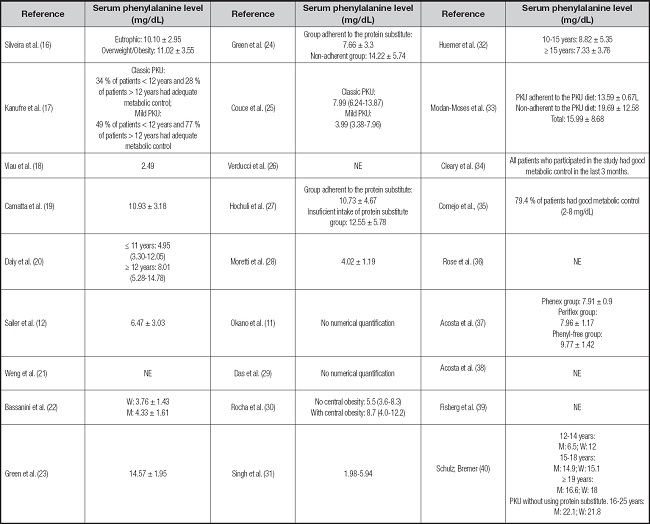
PKU: phenylketonuria; NE: not evaluated, not present; M: men; W: women.
Serum phenylalanine results were standardized in mg/dL.
On the other hand, the blood phenylalanine level (Table IV) was described by most studies and ranged from 1.98 to 22.1 mg/dL. The metabolic control of the disease is classified as adequate when blood phenylalanine values are between 2-6 mg/dL for the age group from 0 to 12 years and between 2 and 10 mg/dL for those older than 12 years (3). Some studies evidenced adequate metabolic control in most of the sample analyzed (11,19,20,24,29,32,34,35) while others observed levels above reference values (23,27,37,40). Studies that compared different groups of individuals with PKU mostly found no statistically significant difference between the groups (16,33) and only one article found different results according to type of PKU, with unsatisfactory (high) metabolic control in classical PKU and adequate control in mild PKU (17).
DISCUSSION
The study presented here investigated the evidence of dietary intake of individuals with PKU and the metabolic control of the disease and, demonstrated that individuals tend to have low energy, protein, and fiber intake, adequate lipid intake, and high glucose intake, with elevated blood phenylalanine levels.
The most commonly used instrument to assess food intake in this study was the three-day food record. Only one study used the 24-hour food recall method plus the Multiple Pass method. This method consists of five steps, starting with the participant’s fast report of all foods and beverages consumed in an uninterrupted manner, and at the end of the fast report, the respondent is asked if he or she remembers any other food or beverage that he or she did not report, with a listing of commonly forgotten foods by the interviewer. At the end of this report, the participant is asked about the type, time and place of each meal. Then a detailed breakdown of the food is requested, including, for example, preparation method, quantities, sizes, and home measures, as well as information on the addition of other foods. The interview is concluded with a complete review of the foods, with the interviewer listing the report to the interviewee, encouraging the reporting of possibly forgotten and/or omitted foods (41).
The energy intake of individuals with PKU is lower than that of healthy groups (18,31,37,39,40). The lower energy intake may be related to low food intake, failure to record some foods (37), the socioeconomic situation of the family, food neophobia and/or fear of reprisal in the case of food consumption higher than the recommendation, or even omission by consumption of foods considered prohibited, and also the relationship and trust of the patient in his team of health professionals.
So far, only one study compared the resting energy expenditure of adolescents with PKU with healthy adolescents and found no statistically significant differences between them (42). As well, only one study evaluated the resting energy expenditure of children and adults with PKU and found no statistically significant difference with their respective control groups (43). More studies are needed to confirm that PKU does not alter the energy expenditure of individuals.
In children with PKU, food intake is based on a smaller number of foods, offering little dietary variety (20). Thus, the use of protein substitutes (bread, noodles, cookies, cake mixes, low-protein cereal bars, and animal milk substitutes, for example) can contribute to reduced natural protein intake and provide 30 to 50 % of energy requirements (20,28), improving diet variety, nutritional management (44), food and nutrition security (45) and metabolic control of the disease, as protein catabolism leads to increased phenylalanine concentrations in the blood (20). However, on the other hand, uncontrolled use of protein substitutes may contribute to obesity (28,44) since there is still more attention on taste and presentation than on nutritional composition (4,28).
A considerable number of papers demonstrate that the protein intake of individuals with PKU is insufficient (12, 26-29). In PKU patients, a well-defined dietary treatment with phenylalanine-free L-amino acid blended formulas and special low-protein foods provides natural protein restriction, adequate protein intake, and adequate growth and development (6,46).
Protein recommendations for individuals with PKU stem from protein recommendations for healthy individuals, additional factors that may influence protein utilization in PKU such as assimilation of phenylalanine-free amino acids compared to natural (intact) protein, growth of individuals with PKU, and studies investigating metabolic control with different dosages of phenylalanine-free amino acids (4). In consensus, it is recommended to provide 20 % additional amino acids daily, on ideal body weight, to compensate for losses of undigestible amino acids and an additional 20 % to optimize the effect of amino acids on blood phenylalanine control (8,47).
Studies have shown that amino acid mixtures show differences in intestinal absorption rate compared to natural protein, with faster absorption peaks, steeper reductions in blood concentration, and greater nitrogen losses (46). Glycomacropeptide (GMP), a protein derived from cheese whey, rich in threonine and isoleucine and almost free of phenylalanine, tyrosine and tryptophan, has been used, with supplementation of other essential amino acids without phenylalanine, in the therapeutic diet of individuals with PKU (6,48). Studies with this protein demonstrate slower absorption, lower L-amino acid degradation, better protein retention compared to phenylalanine-free amino acids, improved palatability (49,50), immunomodulatory role (50,51) and improve the biodiversity of the intestinal flora by presenting a prebiotic role, since its structure has extensive glycosylation with sugars (sialic acid, galactosyl and N-acetylgalactosamine), which are substrates for some beneficial bacteria such as Lactobacillus and Bifidobacteria (6,50,51).
With reduced protein intake, individuals also tend to reduce their fat intake and energy needs can be met by overconsumption of carbohydrates (12,25,52) with a high glycemic index (28). The starch sources of low-protein foods of bread and pasta are usually derived from isolated starch from wheat, corn, and rice. Isolated starches are refined, with different physiological properties compared to complex forms of starch, and foods containing them may have a higher glycemic index than those made with wheat flour. Also, the high intake of sugar in sweet drinks is also problematic. On the other hand, many “sugar-free” beverages are unsuitable for individuals with PKU because they contain aspartame, a sweetener that is a source of phenylalanine, limiting the choice of foods consumed (20).
Insulin resistance, as measured by HOMA-IR (Homeostasis Model Assessment Insulin Resistance), is higher in individuals with PKU compared to healthy controls (25). This fact may be related to metabolic changes caused by prolonged intake of high levels of carbohydrates, especially if accompanied by excess energy intake (25). In addition, higher triglyceride rates have been reported in individuals with PKU, suggesting an association of serum lipids with the quality of the carbohydrate consumed (20,28,52).
On the other hand, it is known that carbohydrate intake stimulates insulin secretion that culminates in protein synthesis and increased amino acid transport into cells (53). In this regard, a study showed that postprandial net protein absorption improved by 5 % and nitrogen retention by 14 % when carbohydrates were ingested along with protein (54). Therefore, providing the phenylalanine-free amino acid formula along with carbohydrates, especially of good quality, to individuals with PKU optimizes the nutrients offered.
The consumption of fiber in the population with PKU is still poorly studied. Different sources of fiber interfere with the gut microbiome, increasing or decreasing the risk of chronic diseases such as inflammatory bowel disease and obesity (20). Gene richness of the fecal microbiome is positively correlated with consumption of fruits, vegetables, polyphenols, and prebiotics (6,55). Fruits and vegetables low in phenylalanine (≤ 75 mg/100 g), except potatoes, do not impact the control of phenylalanine levels and should be encouraged in a low phenylalanine diet as a source of beneficial fiber (56). On the other hand, cereal and whole grain fibers, associated with a lower risk of cardiometabolic disease and colorectal cancer should be excluded from a phenylalanine-restricted diet (20).
Lipid intake by individuals with PKU is within the RDA (12,18,20,22-24,26-29,37,40). The main source of fats in the diet of individuals with PKU are vegetable oils, butter, margarine, and small amounts of heavy cream. And, as in vegetarian diets, vegetable fats provide mostly, polyunsaturated fatty acids, mainly linoleic acid (40), which reflects in lower total/LDL cholesterol rates (20,52). Also, elevated phenylalanine levels are associated with impaired cholesterol synthesis due to dysregulated expression of 3-hydroxy-3-methylglutaryl-CoA reductase and inhibition of mevalonate 5-pyrophosphate decarboxylase, in addition to high consumption of acetyl CoA to synthesize phenylacetylglutamine, causing hypocholesterolemia (52,53).
Low plasma concentrations of linolenic acid, arachidonic acid (AA), docosahexaenoic acid (DHA), and eicosapentaenoic acid (EPA) have been found in PKU patients (57,58). Diet deficient in essential fatty acids and the accumulation of toxic metabolites can affect the enzymes of lipid metabolism, causing increased oxidative stress, lipid peroxidation, and inflammation (59). In these cases, supplementation of essential fatty acids may be necessary, since these fatty acids play an important role as a constituent of cell membranes, cognitive functioning and visual development (53).
The results on the dietary intake of individuals with PKU prompt reflection on the factors that influence food choice, which are complex and include access to adequate food, socioeconomic components, nutritional knowledge and monitoring, psychological and cultural aspects. In addition, parents’ eating habits are an important factor influencing their children’s food choice. Food preferences and aversions develop mainly during the first years of life, resulting in stable eating patterns, which imposes the need for continuous actions of food and nutrition education for this population, starting as early as possible.
Data on the metabolic control of PKU in different individuals show the difficulty in maintaining blood phenylalanine values at adequate levels. The mechanism by which elevated phenylalanine causes neurotoxicity in PKU is unclear. Possible explanations include reduced amino acid transport to the brain and subsequent reduced protein production and enzyme activities, particularly enzymes involved in neurotransmitter synthesis. Elevated blood phenylalanine competes with the transport of large neutral amino acids (including tyrosine and tryptophan) to the brain, causing an overall reduction in the levels of these amino acids in the brain (60). Although neurotoxicity is the hallmark of PKU pathology, there is also an effect of hyperphenylalanemia in the liver, the primary site of PAH enzyme expression. Transcriptome and proteomics data have shown alterations in a large number of liver genes, suggesting impairment of metabolic pathways of energy substrate utilization, but with reversible changes after adjustment of blood phenylalanine levels (59,60).
The very limited diet, the need to maintain strict eating routines, and the preparation and adequate consumption of the phenylalanine-free amino acid formula may require management of 19 hours per week (17), and in the scarcity of time for planning activities, these factors contribute to inadequate metabolic control.
This is the first study we know of that has provided an overview of the dietary intake and blood phenylalanine levels of different individuals with PKU and followed most of the steps of a systematic review. However, this study was unable to characterize dietary intake in each life cycle, as most studies did not separate individuals in their analyses.
CONCLUSION
This study showed that individuals with PKU tend to have low energy, protein and fiber intake, adequate lipid intake, and high glucose intake. The metabolic control of the disease in individuals is still a challenge in all countries. The search for drug and non-drug strategies to control blood phenylalanine levels in individuals with PKU remains an urgent need.

