










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.21 no.4 abr. 2004
| Manifestaciones óseas de la enfermedad de C. CAMPO LÓPEZ, J. R. CALABUIG ALBORCH, J. AGUILAR JIMÉNEZ, Servicio de Medicina Interna. Hospital Universitario La Fe. Valencia
|
| SKELETAL MANIFESTATIONS OF GAUCHER´S DISEASE. A REPORT OF TWO CASES
| ||
| RESUMEN La enfermedad de Gaucher es la enfermedad hereditaria de depósito lisosomal más frecuente. Se caracteriza por una deficiencia de la enzima glucocerebrosidasa que conduce a la acumulación de substrato en el interior de los lisosomas de los macrófagos. Se clasifica en tres tipos según exista afectación de sistema nervioso central (tipos 2 y 3) o no (tipo 1). Es una patología multisistémica y en la mayoría de pacientes se aprecia hepatoesplenomegalia, anemia y trombopenia. La afectación esquelética también es importante y a menudo constituye el aspecto más discapacitante. Presentamos dos casos de enfermedad de Gaucher con manifestaciones óseas y realizamos una revisión de la literatura.PALABRAS CLAVE: Enfermedad de Gaucher. Médula ósea. Hueso. Complicaciones. | ABSTRACT Gaucher'disease is the most frequent hereditary lysosomal deposit storage disorder. It is characterized by a deficiency of the enzyme glucocerebrosidase that leads to an accumulation of substrate in the interior of the macrophage lysosomes. It is classified in three types, according to the presence of central nervous system involvement (type 2 and 3) or not (type 1). It is a multisystemic disease and in the majority of patients there is hepatosplenomegaly, anemia and thrombocytopenia. Skeletal involvement is also important and it is frequently the most disabling manifestation. We present two cases of Gaucher´s with skeletal manifestations and we review the literature.KEY WORDS: Gaucher's disease. Bone marrow. Bone. Complications. |
Campo López C, Calabuig Alborch JR, Aguilar Jiménez J, Alonso Estellés R. Manifestaciones óseas de la enfermedad de Gaucher. A propósito de dos casos. An Med Interna (Madrid) 2004; 21: 179-189.
Trabajo aceptado: 14 de octubre de 2003
Correspondencia: Cristina Campo López. Servicio de Medicina Interna. Hosital Universitario La Fe. Avda. de Campanar, 21. 46009 Valencia
INTRODUCCIÓN
La enfermedad de Gaucher (EG), descrita en 1882 como esplenomegalia no leucémica, es la más frecuente de las anomalías hereditarias de carácter lisosomal (1,2). Se trata de un trastorno genético de herencia autosómica recesiva producido en la mayoría de los casos por mutaciones que afectan al gen de la beta glucocerebrosidasa ácida (GBA) localizado en el brazo largo del cromosoma 1 (2). Dicho defecto produce una enzima deficiente que no es capaz de degradar su sustrato correspondiente con el consiguiente acúmulo de glucosilceramida y otros esfingolípidos en los lisosomas de los macrófagos del sistema reticular histiocítico. Estas células constituyen las llamadas células de Gaucher (3). La correlación fenotipo-genotipo es poco significativa en esta enfermedad y genotipos idénticos pueden manifestarse por cuadros clínicos dispares (4). Clásicamente se clasifica en tres tipos según el SNC esté indemne (tipo1 no neuropático) o no (tipo 2 neuropático agudo y tipo 3 neuropático subagudo). El tipo 1 es el más frecuente y presenta una elevada prevalencia en judíos Ashkenazi. Muestra una marcada heterogeneidad clínica predominando la esplenomegalia, anemia, trombocitopenia y afectación ósea (5). Aunque las complicaciones esqueléticas se diagnostican y evalúan peor, a menudo constituyen el aspecto más discapacitante de la EG. La progresiva infiltración de la médula ósea por macrófagos cargados de glucocerebrósido conduce a fallo en la remodelación, osteopenia, fracturas patológicas, osteoesclerosis, osteonecrosis, crisis óseas y retraso en el crecimiento (6). En consecuencia los pacientes experimentan dolor crónico, deformidad e incapacidad. Desde su descripción se han producido importantes avances en el conocimiento de la enfermedad y en su pronóstico al disponer desde 1990 de un tratamiento eficaz mediante la infusión periódica de la enzima funcionalmente activa (TRE). Dicho tratamiento ha demostrado respuestas viscerales y hematológicas notables junto a una mejora de la mineralización ósea y de las manifestaciones esqueléticas (7).
CASOS APORTADOS
Presentamos dos casos de EG tipo1 con distintas manifestaciones óseas a lo largo de su evolución.
Caso 1: Varón nacido de embarazo y parto normal, diagnosticado a los 9 años de enfermedad de Gaucher tipo 1 tras ser estudiado por anorexia importante, palidez, hepatoesplenomegalia, anemia y trombopenia. Esplenectomizado con éxito a los 19 años por pancitopenia. Genotipo N370S/N370S. Dolores óseos aislados durante la infancia. Diagnosticado de aplastamiento vertebral T11 y L4 a los 28 años con paraparesia por compresión (Fig. 1). Otros hallazgos radiológicos de enfermedad de Gaucher se muestran en la tabla I. Intervenido posteriormente mediante corporectomía T11 con relleno de cemento e injerto, y en un segundo tiempo artrodesis posterior desde T7 a L5 con descompresión de L4. Buena evolución postquirúrgica. En tratamiento enzimático sustitutivo desde 1998.

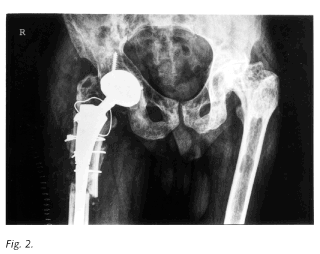
Caso 2: Varón diagnosticado de enfermedad de Gaucher tipo 1 a los 7 años tras ser estudiado por astenia, anorexia, hepatoesplenomegalia, pancitopenia y dolores óseos. Esplenectomizado a los 11 años por pancitopenia severa. Genotipo N370S/N370S. Múltiples episodios de dolor óseo localizado y autolimitado durante la infancia tratados de forma sintomática. Osteoesclerosis de cadera izquierda asintomática desde los 15 años. Prótesis de cadera derecha por necrosis avascular de cabeza femoral a los 40 años (Fig. 2).Otros hallazgos radiológicos de enfermedad de Gaucher (Tabla I). Recambio de la prótesis tres años después por aflojamiento aséptico. Buena evolución posterior. Ha rechazado en varias ocasiones el tratamiento enzimático sustitutivo por interferencia en su estilo de vida e independencia.
DISCUSIÓN
En el hueso normal los triglicéridos son el componente lipídico dominante (mas del 80% del contenido total de lípidos) junto con una pequeña cantidad de glucocerebrósidos en la médula. En la EG se produce un aumento de glucocerebrósidos con la consiguiente disminución del porcentaje de triglicéridos, disminuyendo el contenido total lipídico medular (8).
Se cree que la causa fisiopatológica primaria de la afectación ósea en la enfermedad de Gaucher es la infiltración y acumulación de macrófagos anormales en la médula ósea y liberación local por estas células de sustancias que favorecen la resorción ósea (6,9). Existe controversia en el papel de la esplenectomía en el curso de la infiltración ósea y la mayoría de autores creen que exacerba la afectación medular al eliminar una zona de almacenamiento de células patológicas (10).Se estima que más del 80% de los pacientes con EG tienen cierto grado de afectación esquelética y que más de la mitad de ellos experimentan complicaciones graves. Las complicaciones clínicas resultantes incluyen dolor, deformidad e incapacidad que pueden requerir tratamientos médicos e intervenciones quirúrgicas de cierto riesgo y que influyen negativamente en la calidad de vida del paciente (7,11).
La extensión de la afectación esquelética varía de unos pacientes a otros y no puede predecirse a partir de la edad del individuo, del déficit de glucocerebrosisdasa, de la afectación de los sistemas visceral y hematológico, o del genotipo. Los hallazgos radiológicos son variables dependiendo de la cantidad de células de Gaucher acumuladas en el sistema reticulo endotelial (6,7,12).
Actualmente se dispone de numerosas técnicas de imagen para la evaluación del hueso cortical, trabecular y de la médula ósea. La radiografía simple es insensible para detectar cambios óseos precoces pero es eficaz para determinar el grosor cortical y permite evaluar las articulaciones, alteraciones en la remodelación y lesiones escleróticas. Es el método más accesible y barato pero al tratarse de radiación ionizante entraña ciertos riesgos sobre todo en niños (8,13). La RM es el método más sensible e inocuo para la evaluación de la infiltración medular ya que la alteración en la distribución de sus distintos componentes provoca cambios precoces en la intensidad de señal. Actualmente se considera un método de elección tanto para el diagnóstico de la enfermedad como para el seguimiento tras tratamiento enzimático (14,15,24). Otras técnicas de imagen como la tomografía computarizada, la RM de desplazamiento químico cuantitativo (MRQCSI) y la absorciometría de rayos X de energía dual (DEXA) son también útiles, pero su accesibilidad es limitada (7,16,19).
En los estadios más precoces de la enfermedad, la infiltración de células de Gaucher comienza en las vértebras propagándose posteriormente hacia pelvis, fémures y tibias. La osteopenia puede llegar a ser importante, localizada o difusa y afectando tanto a hueso trabecular como cortical (17).
Las vertebras planas aparecen frecuentemente en el segmento dorsal por necrosis y fracturas por compresión junto con ensanchamiento del espacio discal. Pueden producir deformidad con cifosis y escoliosis y provocar dolor y déficit neurológico que requiera reducción o intervención quirúrgica, como en nuestro primer caso (6,18). La deformidad en matraz de Erlenmeyer es característica, aunque no patognomónica, de esta enfermedad. Se produce por presión del conglomerado de células de Gaucher medulares sobre el endostio en los huesos tubulares, fundamentalmente el fémur distal y tibia proximal, e inadecuada remodelación de la zona metafisaria (17,19). En nuestro segundo caso se pudo observar esta deformidad también en el humero derecho, localización menos frecuente.
En ocasiones, también en nuestro caso 2, se observan áreas osteolíticas múltiples y osteoesclerosis como resultado de una hiperactividad osteoblástica y osteoclástica que se asocia a infartos óseos (6,9).
La necrosis aséptica suele aparecer en extremos de huesos de carga sobre todo en las cabezas femorales, pero también se ha visto en humeros y huesos de manos y tobillos. Se debe a infartos crónicos producidos por oclusión arteriolar y episodios de vasoespasmo y trombosis (19,20). El colapso articular obliga a practicar cirugía protésica, como en el segundo paciente comentado. Probablemente constituye la complicación ósea más incapacitante de la EG.
Las crisis óseas son episodios frecuentes de dolor agudo localizado en huesos largos y acompañado de fiebre y síntomas sistémicos. Son causadas por infartos óseos agudos y más frecuentes en las dos primeras décadas de la vida. El diagnóstico diferencial con la osteomielitis se realiza mediante gammagrafía, que no capta en estos casos. Suelen ser autolimitadas y se resuelven en dos o tres semanas con tratamiento sintomático (8,21). En nuestro caso 1 fueron aisladas y se resolvieron sin problemas pero en el caso 2 fueron múltiples y obligaron a utilizar analgésicos y AINES potentes que ocasionaron hemorragia digestiva aguda con el consiguiente ingreso hospitalario.
Los efectos clínicos de la TRE, como la reducción del dolor óseo, pueden observarse a los 3 ó 4 meses del inicio de la terapia con la consiguiente mejora de la calidad de vida del paciente. Los efectos sobre la fisiopatología del hueso se observan de forma más tardía. Se ha detectado respuesta medular ósea a los 6 meses del tratamiento con disminución de los niveles de glucocerebrósido e incremento en el contenido de triglicéridos. La respuesta en los compartimentos cortical y trabecular del esqueleto sólo se ha detectado después del tratamiento a largo plazo utilizando dosis de 60 U por kg de peso cada 2 semanas (7,22).
Pese a sus múltiples ventajas, este tratamiento obliga a acudir frecuentemente a un centro hospitalario lo que interfiere en la independencia y estilo de vida del sujeto por lo que algunos, como el comentado en segundo lugar, se niegan a recibirlo. Aunque con un sólo paciente, ya existe un trabajo publicado en España de tratamiento domiciliario (23) con buenos resultados.
Para evitar la progresión a enfermedad ósea irreversible es importante iniciar la TRE lo antes posible. En niños un inicio terapéutico precoz puede lograr que obtengan una masa esquelética normal.
Bibliografía
1. Gaucher PCE. De l'épithélioma primitif de la rate, hyperthrophie idiopathique de la rate sans leucémie. Doctoral thesis. París, 1882. [ Links ]
2. Barranger JA, Rice E. An overview of Gaucher disease. Gaucher Clinical Perspectives 1993; 1: 1-4. [ Links ]
3. Beutler E. Gaucher disease: New molecular approaches to diagnosis and treatment. Science 1992; 256: 794-799. [ Links ]
4. Mistry PK. Genotype/phenotipe correlations in Gaucher's disease. Lancet 1995; 346: 982-983. [ Links ]
5. Perez-Calvo JI, Pastores GM, Isol AL, Giraldo P, Bueno-Gomez JM. Enfoque terapéutico de la enfermedad de Gaucher tipo 1. Sangre 1994; 39: 39-44. [ Links ]
6. Pastores GM, Einhorm TA. Skeletal complications of Gaucher disease: pathophysiology, evaluations and treatment. Semin Hematol 1995; 32 Suppl 1: 20-27. [ Links ]
7. Rosenthal DI, Doppelt SH, Mankin HJ, Dambrosia JM, Xavier RJ, McKusick KA, et al. Enzyme replacement therapy for Gaucher's disease: skeletal responses to macrophage-targeted glucocerebrosidase. Pediatrics 1995; 96: 629-637. [ Links ]
8. Espin JA, McPherson EJ. Treatment of bone complications in patients with Gaucher disease. Gaucher Clin Perspect 1994; 2: 7-11. [ Links ]
9. Adarraga MD, Fernández de la Puebla R, Jiménez JA, Pocoví M, Zambrana JL, Pérez F. Enfermedad de Gaucher. Aportación de 4 casos. Rev Clin Esp 2002; 202: 635-637. [ Links ]
10. Rodriguez SW, Rosenthal DI, Barton NW, Zurakowski D, Mankin HJ. Risk factors for osteonecrosis in patients with type 1 Gaucher's disease. [ Links ]
11. Mankin HJ, Rosenthal DI, Xavier R. Gaucher disease: new approaches to an anciert disease. J Bone Join Surg Am 2001; 83: 748- 762. [ Links ]
12. Wenstrup RJ, Roca-Espiau M, Weinreb NJ, Bembi B. Skeletal aspects of Gaucher disease: a review. Br J Radiol 2002; 75 Suppl 1: A2-12. [ Links ]
13. Rademakers RP. Radiologic evaluation of Gaucher bone disease. Semin Hematol 1995; 32 (Supl. 1): 14-19. [ Links ]
14. Maas M, Poll LW, Terk MR. Imaging and quantifying skeltal involvement in Gaucher disease. Br J Radiol 2002; 75 (Supl. 1): A13-24. [ Links ]
15. Poll LW, Koch JA, vom Dahl S, Willers R, Scherer A, Boerner D, et al. Magnetic resonance imaging of bone marrow changes in Gaucher disease during enzyme replacement therapy: first German long-term results. Skeletal Radiol 2001; 30: 496-503. [ Links ]
16. Maas M, Hollak CE, Akkerman EM, Aerts JM, Stoker J, Den Heeten GJ. Quantification of skeletal involvement in adults with type 1 Gaucher's disease: fat fraction measured by Dixon quantitative chemical shift imaging as a valid parameter. Am J Roentgenol 2002; 179: 961-965. [ Links ]
17. Einhorn TA. Managing orthopedic manifestations of Gaucher disease. Pawling, NY: Caduceus Medical Publishers 1993; 1-4. [ Links ]
18. Minchew JT, Dawson EG. The spine and Gaucher disease. Gaucher Clin Perspect 1994; 2: 1-6. [ Links ]
19. Hainaux B. Gaucher disease: plain radiography, US, CT and MR diagnosis of lung, bone and liver lesions. Pediatr Radiol 1992; 22: 78. [ Links ]
20. Mankin HJ, Doppelt SH, Rosenberg AE, Barranger JA. Metabolic bone disease in patients with Gaucher's disease. In: Avioli LV, Krane SM, editors. Metabolic bone disease and clinically related disorders (2nd edn). Philadelphia PA: WB Saunders Company 1990; 730-752. [ Links ]
21. Yosipovitch Z, Katz K. Bone crisis in Gaucher disease - an update. Isr J Med Sci 1990; 26: 593-595. [ Links ]
22. Weinreb NJ, Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, et al. Effectiveness of enzime replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med 2002; 113: 112-119. [ Links ]
23. Pérez-Calvo JI, Giraldo P. Tratamiento domiciliario de la enfermedad de Gaucher tipo 1 en España. Med Clin (Barc) 2002; 119: 756. [ Links ]
24. Pérez-Calvo JI, Giraldo P, Rubio-Félix D, Giralt M, Pocoví M. Estudio de calidad del diagnóstico y seguimiento de los trastornos esqueléticos asociados a la enfermedad de Gaucher tipo 1 en España. An Med Interna (Madrid) 2003; 20: 607-611. [ Links ]

