










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.23 no.5 may. 2006
CARTAS AL DIRECTOR
Tumor de origen desconocido como presentación de rabdomiosarcoma embrionario
Embryonal rhabdomyosarcoma presenting as tumor of unknown origen
A. B. Mencías Hurtado; M. C. Maeso Fortuna; M. R. Hernández Kauffman; M. Morales González
Servicios de Rehabilitación, Anatomía Patológica y Oncología Médica. Hospital Universitario Nuestra Señora de Candelaria. Santa Cruz de Tenerife.
Sr. Director:
El rabdomiosarcoma infantil, es un tumor maligno del tejido blando de origen musculoesquelético, que representa aproximadamente el 3,5% de los casos de cáncer en niños de 0 a 14 años de edad, y un 2% entre adolescentes y adultos jóvenes entre 15 a 29 años de edad. La media de edad al diagnóstico son 4 años (1). Las localizaciones primarias más comunes donde aparece son cabeza y cuello (tejido parameníngeo, órbita, faringe, senos paranasales); conducto genitourinario y extremidades. Otros sitios primarios menos comunes incluyen la región intratorácica, el aparato digestivo (incluyendo hígado y vías biliares) y la zona perineal (2). La mayoría de casos de rabdomiosarcoma se presentan de forma esporádica. Sin embargo, en una pequeña porción se han detectado mutaciones específicas (3). A continuación se presenta un caso inusual por la edad y la forma de presentación al diagnóstico.
Varón de 23 años sin antecedentes personales ni familiares de interés, que ingresa para estudio por fiebre intermitente de 38-39 ºC, astenia progresiva y lumbalgia de características mecánicas, de 2 meses de evolución.
La exploración física muestra un varón consciente y orientado; con intensa palidez cutáneo mucosa; microadenopatías laterocervicales bilaterales e inguinales móviles, no dolorosas, y dos nódulos cutáneos a nivel axilar izquierdo, duros y no dolorosos; siendo el resto de la exploración normal.
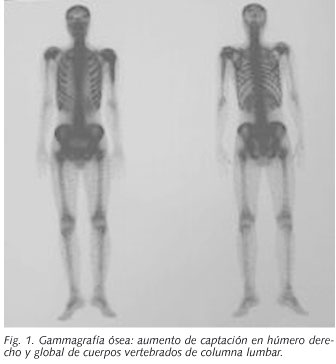
Los resultados más importantes de las pruebas complementarias realizadas fueron los siguientes. En la analítica de sangre destacan: VSG: 82; Hb: 7,2; GOT: 67; GPT: 56; GGT: 113; LDH: 2667; hierro: 45 y ferritina: 3826. Una TAC tóraco-abdominal, muestra adenopatías axilares bilaterales y nódulos subpleurales múltiples. La RMN de columna vertebral completa informó de la presencia de un proceso infiltrativo a nivel de estructuras aracnoideas de la médula en su totalidad. La gammagrafía ósea mostró aumento de captación del trazador en húmero derecho sugestivo de lesión ósea metabólicamente activa y aumento de captación global de los cuerpos vertebrales de columna lumbar. (Fig. 1)
Se realizó punción lumbar, obteniéndose líquido xantocrómico con citología negativa para células malignas. Una punción-aspiración con aguja fina, de unos de los nódulos subcutáneos fue positivo para células tumorales malignas, sugestivas de tumor indiferenciado de células pequeñas. Ante ello, se realiza exéresis de uno de los nódulos, mostrando el estudio con hematoxilina-eosina una prolifereación tumoral maligna de células pequeñas con alto grado de anaplasia, compartimentada en septos fibrosos gruesos que delimitan sábanas tumorales con centros que tienden a la necrosis, infiltrando los tejidos blandos circundantes sin que se reconozca estructura ganglionar linfoide subyacente. La celularidad tumoral muestra discreta cantidad de gotas de glucógeno intracitoplásmico y nunca evidencia estriaciones transversales o diferenciación de cualquier otra estructura.
Desde el punto de vista inmunofenotípico, la tumoración es positiva para vicentina (fuerte y difusamente) y la desmina (más débil y focal). Las citoqueratinas y otros marcadores epiteliales, sinaptofisina, S-100, Hmb45, CD99 y LCA fueron negativos. También hubo negatividad para actina específica de músculo liso, CD31 y CD34. El diagnóstico patológico fue de sarcoma indiferenciado de células pequeñas de patrón histológico e inmunohistoquímico concordante con rabdomiosarcoma embrionario. Se realizó biopsia de médula ósea, observándose masiva infiltración de la misma, por una población celular idéntica a la anteriormente descrita. Una TAC de senos paranasales fue normal. Tras el diagnóstico se inicia tratamiento quimioterápico, según esquema VAC (vincristina, actinomicina D, ciclofosfamida) y metrotrexate intratecal; junto a tratamiento antitérmico, morfina oral y transfusiones de concentrados de hematíes. Al mes se constata progresión de las metástasis pulmonares y se cambia al esquema MAID (mesna-ifosfamida, adriamicina y dacarbacina), obteniéndose una mejoría clínica, con desaparición de la fiebre, del dolor (no precisó más morfina) y de la anemia (no precisando más transfusiones). A los 8 meses del diagnóstico, se objetiva una progresión importante de las metástasis pulmonares, instaurándose tratamiento paliativo. El paciente fallece dos meses después.
El presente caso tiene las particularidades de la edad de presentación tardía y el de debutar como una enfermedad diseminada, sin masa tumoral principal. La media de edad al diagnóstico son los 4 años y la mayoría de los pacientes debutan con una masa tumoral principal. De las diferentes formas histológicas de rabdomiosarcoma, el tipo embrionario es la forma más frecuente. El 25% de los están diseminados al diagnóstico y en un 10% la médula ósea esta infiltrada en el momento del diagnóstico.
En el presente caso, inicialmente se sospecha una neoplasia hematológica. Sin embargo, la ausencia de blastos en sangre periférica y de trombocitopenia hace poco probable el síndrome mieloproliferativo. La citología del nódulo cutáneo, pudiera corresponder a un linfoma de linfocitos pequeños, pero la edad del paciente y lo agresivo de la clínica hace improbable este diagnóstico, ya que este tipo de linfomas son de curso indolente. Existe un caso similar descrito en un paciente varón de 16 años, con fiebre, lumbalgia, astenia y estudio de RMN de columna vertebral, que mostraba infiltración tumoral de la médula ósea de cuerpos vertebrales dorsales y lumbares. El estudio inmunohistoquímico de la biopsia de médula ósea, fue positivo para desmina y vicentina y presentó traslocación t (2;13) (q35;q14) estableciendo el diagnóstico de rabdomiosarcoma alveolar (4). Existen casos descritos de rabdomiosarcoma alveolar, simulando al diagnóstico de leucemia aguda o un linfoma de alto grado (5). Pese a su escasa frecuencia, habría que tener al rabdomiosarcoma entre los posibles diagnósticos diferenciales, ante la presencia de infiltración medular en pacientes jóvenes.
Bibliografía
1. Ries LA, Kosary CL, Hankey BF, et al, eds: SEER Cancer Statistics Review, 1973-1996. Bethesda, Md: National Cancer Institute, 1999.
2. Crist W, Gehan EA, Ragab AH, et al. The Third Intergroup Rhabdomyosarcoma Study. J Clin Oncology 1995; 13: 610-630.
3. Gurney JG, Young JL, Roffers SD, et al. Soft tissue sarcomas. In: Ries LA, Smith MA, Gurney JG, et al., eds:Cancer Incidence and Survival Among Children and Adolescents:United States SEER Program 1975-1995. Bethesda, MD: National Cancer Institute, SEER Program, NIH Pub.No.99-4649, 1999, p. 111-123.
4. Maywald O, Metzgeroth G, Schoch C, et al. Alveolar rhabdomyosarcoma with bone marrow infiltration mimicking haematological neoplasia. Br J Haematol 2002; 119:583.
5. Lugen C, Harshad S, Jen L. Alveolar rhabdomyosarcoma with concurrent metastases to bone marrow and lymph nodes simulating acute hematologic malignancy. J Pediatric Hematol/Oncology 2004; 26: 696-697.

