










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versión impresa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.78 no.6 jun. 2003
COMUNICACIÓN CORTA
CARCINOMA DE CÉLULAS MERKEL PALPEBRAL:
A PROPÓSITO DE UN CASO
MERKEL CELL CARCINOMA OF THE EYELID:
A CASE REPORT
DOMÍNGUEZ POLO A1, CASTILLO LAGUARTA J1, CRISTÓBAL BESCÓS JA1,
RODRÍGUEZ MARCO A2, VÁZQUEZ PULIDO N2
| RESUMEN Objetivo/Método: El carcinoma de células Merkel (CCM) es un tumor maligno cutáneo neuroendocrino primario que procede de las células Merkel; estas células se encuentran en los párpados a lo largo del margen palpebral entre las pestañas. Es un tumor infrecuente pero altamente agresivo y metastatizante. Para su diagnóstico histopatológico puede ser necesario un estudio inmunohistoquímico y ultraestructural. Palabras clave: Células Merkel, carcinoma neuroendocrino, párpado, inmunohistoquímica.
| SUMMARY Purpose/Methods: We report the case of a 73-year-old white female suffering from Merkel cell carcinoma (MCC) of the eyelid with an evolution of two and half months. MCC is a cutaneous neuroendocrine malignant tumor arising from Merkel cells. These cells are common along the eyelid margin in between the eyelashes. It is infrequent but highly aggressive and with potential metastases. Immunohistochemical and ultrastructural studies may be necessary for an histopathological diagnosis. Key words: Merkel cells, neuroendocrine carcinoma, eyelid, immunohistochemistry.
|
Recibido:3/7/02. Aceptado: 22/5/03.
Servicio de Oftalmología. Hospital Clínico Universitario Lozano Blesa. Zaragoza. España.
1 Doctor en Medicina.
2 Licenciado en Medicina.
Comunicación presentada la XI Reunión de la Sociedad Española de Cirugía Plástica Ocular y Orbitaria (Barcelona 2001).
Correspondencia:
Ángel Domínguez Polo
C/. Condes de Aragón, 20, 4.º B
50009 Zaragoza
España
INTRODUCCIÓN
Las células Merkel se encuentran en la piel y membranas mucosas a excepción de la conjuntiva. Fueron descritas por Friedrich Merkel en 1875. Son células neuroendocrinas que inicialmente se describieron como células sensoriales tactiles y en la actualidad se cree que derivan de una célula madre epidérmica común con los queratinocitos (1). Su malignización fue demostrada por primera vez por Tang y Toker en 1978 (2). Toker, en 1972, llamó a este tumor carcinoma trabecular. Actualmente se le denomina CCM o carcinoma cutáneo neuroendocrino.
CASO CLÍNICO
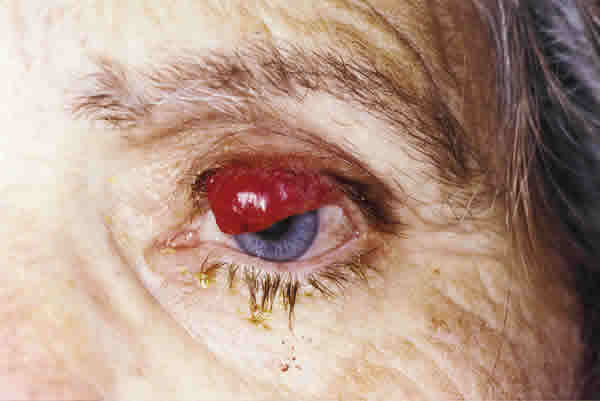
Presentamos el caso de una paciente de 73 años que acudió a la consulta por presentar una tumoración de borde libre palpebral de 2,5 meses de evolución, indolora, sólida, de color rojo oscuro, con telangiectasias superficiales y ulceración focal, que afectaba a más de la mitad del borde libre, con una extensión vertical de pocos milímetros (fig. 1). No existían adenopatías regionales.
Se procedió a la extirpación del tumor y a la reconstrucción del defecto mediante la técnica de Cutler Beard con un colgajo de espesor completo del párpado inferior tallado 5 mm por debajo y paralelo al borde libre palpebral (fig. 2). A las seis semanas se dividió el colgajo para rehacer los párpados, quedando a los tres meses como se muestra en la figura 3.
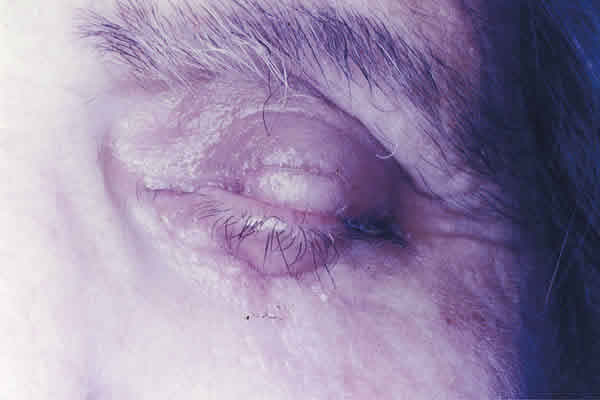
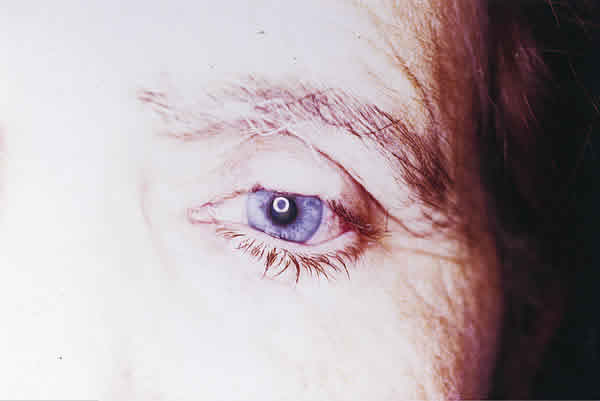
El estudio histopatológico reveló un tumor densamente celular, con un patrón de crecimiento trabecular, compuesto por células redondas de mediana talla con escaso citoplasma y núcleo grande vesiculoso con pequeño nucleolo y alguna figura de mitosis (fig. 4).
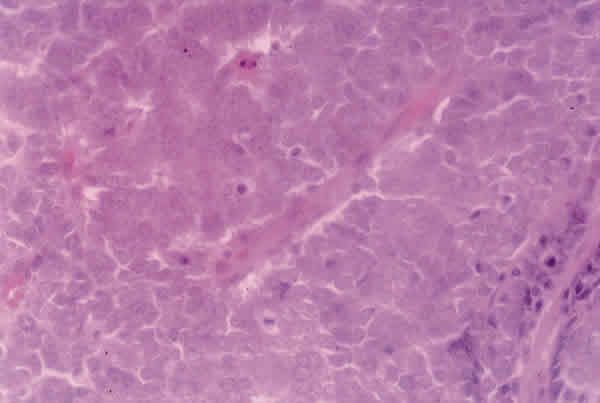
alguna mitosis (H-E 40x).
Se practicó un estudio inmunohistoquímico dando reactividad negativa frente a antígeno común leucocitario y HMB45 y francamente positiva frente a cromogranina (fig. 5), confirmando el diagnóstico de carcinoma maligno de células Merkel.
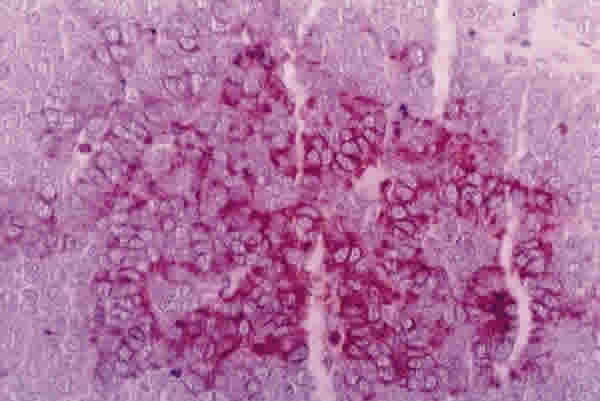
En los tres años y medio de seguimiento, no se han producido recidivas locales ni metástasis.
DISCUSIÓN
El CCM afecta a personas mayores, con predominio de la década de los 60-70 años, sin predilección de sexos, siendo raro en la raza negra. Tiende a aparecer en áreas expuestas de cabeza y cuello (50%) y extremidades superiores. La incidencia en los párpados o región periocular es del 10% con preferencia en el párpado superior. Ocasionalmente puede aparecer en varias localizaciones produciendo una Merkeliomatosis.
Las manifestaciones oftalmológicas consisten en tumores primarios de párpados y metástasis, aunque raras, en órbita, párpados y úvea (3).
Clínicamente se presenta como un nódulo progresivo, indoloro y solitario cerca del margen palpebral, de color rojo fuerte o violáceo con telangiectasias superficiales, de menos de seis meses de evolución. No suele ulcerarse pero, a veces, como en nuestro caso, puede ocurrir. Los bordes están bien definidos pero puede producirse una extensión lateral en los planos profundos.
Es un tumor altamente agresivo que produce recidivas locales en el 25% de los casos y metástasis regional linfática en el 30%. Las metástasis, regionales y a distancia, se desarrollan en los dos primeros años del diagnóstico (piel, hueso, cerebro, hígado y pulmón). En una serie publicada recientemente de 14 casos de CCM palpebral con un seguimiento medio de 3 años, el índice de mortalidad es del 7% (4).
Histopatológicamente se caracteriza por tener un patrón difuso de cordones trabeculares entrelazados, compuestos por células de 15-20 µm de diámetro, de bordes confusos, escaso citoplasma, núcleo redondo prominente con cromatina dispersa y nucleolo indiferenciable. Suelen existir muchas figuras mitóticas y necrosis celular simple y en más de la mitad de los casos invasión vascular y linfática. Periféricamente el tumor está infiltrado por células plasmáticas y linfocitos. En base al microscopio de luz, el CCM puede confundirse con el carcinoma sebáceo mal diferenciado, linfoma, carcinoma de células pequeñas, carcinoma de células basales, tumor carcinoide y metástasis de otras neoplasias neuroendocrinas (1).
El patrón inmunohistoquímico es similar al de otros tumores neuroendocrinos: tinción positiva con anticuerpos para citoqueratina, enolasa neuroespecífica y cromogranina, y tinción negativa para el antígeno común leucocitario.
En ocasiones se necesita un estudio ultraestructural para llegar al dignóstico definitivo, encontrando gránulos neuroscretores citoplásmicos de núcleo denso que contienen cromogranina y agregados filamentarios paranucleares de citoqueratina y/o neurofilamentos que son altamente diagnósticos.
Clínicamente debe diferenciarse del linfoma, carcinoma sebáceo y chalación.
La estrategia terapéutica será individualizada para cada caso, con la colaboración de los Servicios de Radioterapia y Oncología. La agresividad del tratamiento dependerá de la extensión del tumor. Debe realizarse la resección quirúrgica del tumor con márgenes de seguridad de 5 mm. Se aconseja dar radioterapia profiláctica en los tejidos entre el tumor y los ganglios linfáticos primarios (5), así como la exéresis de los ganglios linfáticos afectados y radioterapia local. En caso de extensión orbitaria sin metástasis a distancia, puede requerir la exenteración orbitaria. La quimioterapia tiene una eficacia parcial y se considera paliativa (3).
BIBLIOGRAFÍA
1. Singh AD, Eagle RC, Shields CL, Shields JA. Merkel cell carcinoma of the eyelids. In: Shields JA. Update on malignant ocular tumors. International ophthalmology clinics. Boston: Little, Brown and Company; 1993; 33: 11-17. [ Links ]
2. Tang CK, Toker C. Trabecular carcinoma of the skin: an ultrastructural study. Cancer 1978; 42: 2311-2321. [ Links ]
3. Kivela T, Tarkkanen A. The Merkel cell and associated neoplasms in the eyelids and periocular region. Surv Ophthalmol 1990; 35: 171-187. [ Links ]
4. Peters GB, Meyer DL, Shields JA, Custer PL, Rubin PA, Wojno TH et al. Management and prognosis of Merkel cell carcinoma of the eyelid. Ophthalmology 2001; 108: 1575-1579. [ Links ]
5. Cotlar AM, Gates JO, Gibbs FA. Merkel cell carcinoma: combined surgery and radiation therapy. Am Surg 1986; 52: 159-164. [ Links ]

