










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.96 no.4 Madrid abr. 2004
| POINT OF VIEW |
Gastric carcinogenesis
S. de la Riva, M. Muñoz-Navas and J. J. Sola1
Services of Digestive Diseases and 1Pathology. Clínica Universitaria de Navarra. Pamplona. Navarra, Spain
De la Riva S, Muñoz-Navas M, Sola JJ. Gastric carcinogenesis. Rev Esp Enferm Dig 2003; 96: 265-276.
Recibido: 03-10-03.
Aceptado: 07-10-03.
Correspondencia: Susana de la Riva Onandía. Servicio de Aparato Digestivo. Clínica Universitaria de Navarra. Avda. Pío XII, 36. 31008 Navarra. Telf.: 948 25 54 00.
Fax: 948 29 65 00.
1. INTRODUCTION
Currently, gastric adenocarcinoma is still a serious healthcare concern, and the second most frequent malignancy worldwide (1). Although overall incidence has seemingly decreased, both its frequency and mortality rate show relevant geographic variability. In Eastern countries, Eastern Europe, and South America the incidence of this disease reaches epidemic proportions, and is the first cause of death from malignant growths (2). In contrast, the incidence of this disease is low in other geographic regions (North America, Western Europe, Australia, New Zealand and Israel). Such differences have been partly attributed to both environmental and nutritional factors in terms of food preservation and preparation, and to infectious factors related to the incidence of H. pylori infection in the population. The potential influence of these factors is such that the increasingly decreased incidence and mortality rate of this disease seen from the 1930's to the last 10 years has been attributed to dietary and food preservation changes (3-5).
However, this change in environmental factors does not fully account for known geographic variations or the various progressions and prognoses this condition may entail.
It is nowadays accepted that gastric carcinogenesis is a progressive process in which multiple environmental and epidemiologic, as well as genetic factors play a role, and whose interactions seem to influence not only the devel-opment but also the progression of the disease (6).
2. CARCINOGENIC FACTORS
2.1. Environmental factors
During the past few years, and given that gastric cancer is still one of the most common malignancies worldwide, many studies focused on the identification of environmental risk factors responsible for its wide geographic variability.
Nutritional factors
Populations with a high consumption of salt, smoked food, hot spicy dishes, or fried fat have a high incidence of gastric adenocarcinoma, which suggests a potential role in carcinogenesis (5,7-10).
Nitrite-rich food or water, high carbohydrate ingestion, and scarce access to and consumption of fruits, fresh vegetables, milk, vitamins A, C and E, and selenium seem to augment the risk of gastric cancer (3,5).
Factors classically considered carcinogenic such as tobacco, alcohol or green tea are the subject of still inconsistent results in the literature (6,11).
A progressive increase of gastric cardial adenocarcinomas in industrialized countries versus predominantly distal varieties and virtually no proximal cases in developing countries may be currently highlighted (9,10). Such differences are attributed to a higher incidence of gastroesophageal reflux disease in developed countries in association with diet and increased obesity rates, and to a chronic use of proton pump inhibitors; this results in saliva nitrates becoming nitrites, which favor the condition (12).
H. pylori infection
In the early 1980's the presence of a bacterium that was initially called Campylobacter piloridis (13) and subsequently C. pylori was unveiled in gastric mucosa biopsy samples from patients with gastritis. It is currently designated Helicobacter pylori. These are spiral-shaped bacteria belonging in the gram-negative germs group that use a wide variety of strategies to survive in acid media.
Chronic H. pylori infection leads to chronic gastritis through the activation of a complex network of inflammatory mediators including IL-8, pro-inflammatory cytokines (IL-1, IL-6, TNFα) and immunosuppressive peptides (IL-10) (14).
This chronic inflammation in turn leads to cell cycle changes, which favor epithelial cell replication and thus increase both the apoptotic rate and the release of oxid-izing substances. All this in combination with a depleted antioxidant defense predisposes against gastric carcinogenesis by increasing the chances of DNA mutations (14).
Such cumulative mutations may lead to the development of premalignant lesions and start the process from metaplasia through dysplasia to gastric adenocarcinoma (4,15).
Chronic H. pylori infection is currently considered a Group I carcinogen by the World Health Organization (WHO) (16), and seems to play a relevant role in the development of distal gastric adenocarcinoma.
A number of clinical and epidemiological studies have reported this association, and it is estimated that at least 1% of H. pylori infections may lead to the development of gastric cancer following the aforementioned sequence (17), with risk increasing by 2.7 to 12 times over that of the general population (18). Overall, 8% of gastric tumors are etiologically related to H. pylori infection (19).
Similarly to the incidence of this disease, the population-based rate of H. pylori infection exhibits a lot of geographic variability. Overall, areas with a high risk of gastric adenocarcinoma show high H. pylori infection rates (20,21).
Similarly, a higher incidence of gastric cancer has been reported in low socio-economic status populations. This observation seems to be partly related to nutritional factors such as restricted access to and consumption of fresh fruits and vegetables, and/or higher, earlier-age H. pylori infection rates (22).
In an attempt to explain the higher association of concomitant H. pylori infection with intestinal-type versus diffuse-type adenocarcinoma, two distinct evolution paths have been proposed in relation to gastric cancer histology, with a number of progressive histologic changes occurring in intestinal-type adenocarcinoma but not in diffuse-type adenocarcinoma (23) (Fig. 1).

Epstein-Barr virus (EBV) infection
Discovered by Epstein in 1964, this is an icosahedral herpesvirus containing a double linear DNA strand. It was first put in relation to gastric cancer in 1990, following the detection of its genetic material in patients with gastric cancer by PCR and in situ hibridation (24). This association has been revealed by numerous studies from various geographic regions (25,26).
In contrast with H. pylori, this viral infection has been detected in both the diffuse and intestinal type of gastric cancer (27).
Its carcinogenic mechanism is still unknown. Two carcinogenesis pathways have been suggested according to the presence or absence of concomitant infection by this virus (28).
2.2. Genetic factors
Regarding the influence of genetic factors on the development of gastric adenocarcinoma, this cancer has been detected in families for two or three generations, and members in a family with a history of gastric adenocarcinoma have a two- or even three-fold increased risk when compared to the general population, which implies a potential for familial aggregation (29).
However, studies including members of only one family are limited and results are influenced by environmental factors, since members belonging to a same family usually share the same dietary and environmental conditions (29).
An observation that indirectly points to a potential role of genetic factors in gastric carcinogenesis is this condition's higher association with blood group A versus the remaining blood groups. This association involves predominantly males and the diffuse rather than intestinal histologic type (5,9,29).
2.3. Precursor conditions
A number of histologic changes in the healthy gastric mucosa significantly increase the risk of gastric adenocarcinoma.
Notable among these are:
Chronic atrophic gastritis
A premalignant lesion present in 90% of gastric adenocarcinomas. It usually takes a lot of time until gastric cancer develops. In most studies with a patient follow-up above 10 years, the risk of gastric cancer was of 1 in 150 patients per year, and it went further up to 10% after 15 years of follow-up (30,31).
Its carcinogenic mechanism seems to rely on decreased hydrochloric acid and pepsin secretion, and increased gastric pH, which encourages a proliferation of dietary nitrate-reducing germs. Nitrosamide and nitrosamine formation, together with a number of dietary factors such as excessive salt ingestion or inadequate consumption of fresh vegetables and fruits, may induce DNA mutations in epithelial cells, thus favoring the development and progression of tissue changes such as intestinal metaplasia and dysplasia, which are considered premalignant lesions (4,32).
Pernicious anemia
A condition that exhibits gastric atrophy and increases the risk of gastric cancer (33), even though only 5-10% of such patients will eventually develop it (31).
Partial gastrectomy
Patients with benign conditions undergoing this surgery have an increased risk of gastric adenocarcinoma as of 10 years following the procedure (34). Risk increases by 50 and 70%, respectively, at 15 and 25 years after surgery (31).
Ménétrier’s disease (hypertrophic gastric disease)
The risk of developing gastric cancer from this tissue change is high, and is estimated as 10-15% in some series; however, as this condition is extremely rare in its own right, such transformation percentage is therefore negligible (35).
Adenomatous polyps
Polyps are a common finding at the gastric mucosa. They may be classified into two types:
-Non-neoplastic polyps with no potential for degeneration (hyperplastic, hamartomatous, inflammatory and heterotopic polyps).
-Neoplastic polyps: adenomas. They amount for 15-20% of polyps found at the gastric mucosa. They are potentially neoplastic with an incidence of malignization around 5-15% of tubular adenomas and 15-75% of vil-lous adenomas. Their tendency towards becoming malignant is directly related to polyp size and the presence of dysplasia and its grade (36).
Peptic ulcer
Chances that benign peptic ulcer may transform into a malignancy are still under discussion and opinions vary. Even though most authors refuse this possibility, the role that H. pylori infection seems to play in the process of gastric carcinogenesis should be considered (37).
Barrett’s esophagus
The increased incidence of gastric cardial adenocarcinoma in developed countries seems to be closely related to their increased incidence of gastro-esophageal reflux disease and Barrett’s esophagus (38,39). Wider-scope studies are still needed to determine other factors playing a role in its development, thus allowing to establish whe-ther proximal gastric cancer has a different pathogenesis versus distal gastric cancer.
2.4. Molecular factors
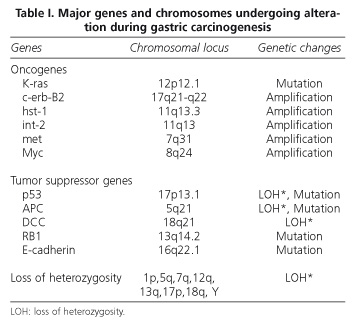
Although evidence that genetic changes play a significant role in the multistep process of gastric carcinogenesis and its progression is available, the huge number of factors analyzed and the various results so far obtained allow no definitive conclusions to be presently drawn (9, 40-42). What seems clear is that the conversion of normal gastric cells into tumor cells is a slow, gradual process in which multiple molecular changes accumulate, and this has been put on a level with the process of colorectal carcinogenesis. This accumulation of genetic changes includes mutations and/or oncogene amplification-overexpression (c-Ki-ras, c-erb-B2, K-sam, hst/int-2, c-met, and c-myc), tumor suppressor gene inactivation (p53, APC, DCC, and RB1), and microsatellite alterations (loss of heterozygosity or microsatellite instability) in one or more chromosomal regions such as 1p, 5q, 7q, 12q, 13q, 17p, 18q, and even chromosome Y (40,43,44) (Table I).
Results from molecular studies on gastric cancer have revealed the potential presence of two different carcinogenesis pathways depending on the adenocarcinoma's histologic type -diffuse or intestinal (45-49). Intestinal-type gastric cancer seems to be related to tissue changes such as intestinal metaplasia within the gastric mucosa, which somewhat resemble the molecular progression of colorectal cancer (47). It follows a process that begins as chronic gastritis and then progresses through atrophic gastritis and intestinal metaplasia to dysplasia (4). On the other hand, the natural history of diffuse gastric cancer lacks such multistep evolution (46,50-52).
3. CURRENT CARCINOGENESIS MODEL
Lauren's histological classification (1965) of gastric cancer into diffuse and intestinal types is most commonly used by a majority of authors (53).
These two types of gastric cancer are probably a reflection not only of morphological differences allowing their classification, but of their varying clinical, epidemiological and pathogenic characteristics.
Therefore, two distinct carcinogenesis pathways have been suggested in relation to these histologic types of gastric cancer, with differing influences of environmental factors and a varying presence and predominance of molecular changes.
As previously stated, the intestinal type seems to develop from a succession of tissue changes in a way that resembles the carcinogenetic pathway of colorectal cancer. Regarding environmental factors, O’Connor et al (54), Watanabe et al (24), and Correa (4) suggested a spiral gastric carcinogenesis model where an accumulation of environmental risk factors entailed a shorter development period, while a reduction of said factors resulted in prolonged development time (Fig. 2).
In addition, similarly to the above-mentioned authors, Yasui et al. (42) collected all molecular factors analyzed and implied in gastric carcinogenesis from the literature in an attempt to put it on a level with the colorectal carcinogenetic process. In view of differences reported between both histologic types of gastric adenocarcinoma, they suggested that two distinct development paths might exist, with different molecular changes being present or predominant (Fig. 3).

What does seem clear is that intestinal-type gastric adenocarcinoma carcinogenesis includes tissue changes that may progress towards malignity and are considered preneoplastic in nature. Such lesions are intestinal metaplasia and epithelial dysplasia.
In contrast, diffuse gastric adenocarcinoma does not seem to evolve along this multistep process and apparently begins in a healthy gastric mucosa that is free of prior tissue changes (34).
4. PREMALIGNANT LESIONS
Premalignant lesions are defined as those tissue changes that may evolve to malignity and are involved in gastric carcinogenesis.
Intestinal metaplasia. Defined as the presence of differentiated epithelium similar to that of the small bowel. It is classified according to both morphologic and histochemical characteristics into:
-Complete intestinal metaplasia (type I).
-Incomplete intestinal metaplasia, either small intestinal (type II) or colonic (type III).
In type-I and type-II intestinal metaplasia goblet cells produce syalomucins, whereas in type-III metaplasia these same cells produce sulfomucins (55,56). Although there seems to be an association between the presence of intestinal metaplasia and gastric cancer. This premalignant lesion is of poor predictive value. Type-I intestinal metaplasia is related to a low incidence of gastric cancer, whereas type-III metaplasia has a 2.7- to 5.8-fold greater risk of gastric cancer development (57,58).
Epithelial dysplasia. Characterized by the presence of a number of histologic changes such as cell atypias with pleomorphism, increased non-differentiated cell numbers, and abnormal crypt and gland layout.
Gastric epithelial dysplasia usually occurs in the setting of atrophic chronic gastritis, and is commonly associated with intestinal metaplasia. Dysplastic areas are commonly found around gastric adenocarcinomas, and are therefore seemingly related -both clinically and pathologically- to gastric cancer. While mild to moderate dysplasia tends to regress or remain stable in most cases, moderate and primarily severe dysplasia is commonly associated with gastric adenocarcinoma development (59) (Fig. 4).

Overall, in 10% of patients epithelial dysplasia may progress towards gastric cancer in a term of 5 to 15 years, but such dysplasia will regress or remain stable in most patients (60).
5. CONCLUSION
Gastric carcinogenesis is a slow and complex process in which multiple environmental and molecular factors seem to play a role. Geographic differences in the incidence, outcome and prognosis of gastric adenocarcinoma seem to be partly related to the varying specific environmental (nutritional and infectious) factors to which populations are exposed. In view of their more than likely involvement, these factors have been thoroughly analyzed and have yielded conclusive results regarding the access to and consumption of selected foods, as well as their preservation and preparation. H. pylori infection amongst the population is another factor involved in the development of gastric cancer, and is nowadays considered a Group I carcinogen by WHO.
Regarding molecular factors, their relevance is increasingly greater for gastric carcinogenesis, and they also play a significant role in the development of this condition. An accumulation of molecular changes seems to influence gastric adenocarcinoma initiation and progression, but its carcinogenic sequence has not been established yet. What seems clear is that such sequence varies according to the histologic type of gastric adenocarcinoma. For this reason, the current categorization of gastric adenocarcinomas by Lauren is considered to reflect not only histologic but also epidemiologic, clinical, and prognostic differences, implying a possibility that two distinct, still unestablished carcinogenesis paths exist.
REFERENCES
1. Roukos DH. Relevant prognostic factors in gastric Cancer. Ann Surg 2000; 232: 719-20. [ Links ]
2. DeVita VT Jr. The war on cancer has a birthday, and a present. J Clin Oncol 1997; 15: 867-9. [ Links ]
3. Forman D. Are nitrates a significant risk factor in human cancer? Cancer Surv 1989; 8: 443-58. [ Links ]
4. Correa P. Human gastric carcinogenesis: a multistep and multifactorial process-First American Cancer Society award lecture on cancer epidemiology and prevention. Cancer Res 1992; 52: 6735-40. [ Links ]
5. Kramer BS, Johnson KA. Other gastrointestinal cancers: stomach, liver. In: Greenwald P, Kramer BS, Weed DL, eds. Cancer prevention and control. New York: Marcel Dekker, 1995. p. 673-94. [ Links ]
6. Kabat G, Ng S, Wynder E. Tobacco, alcohol intake and diet in relation to adenocarcinoma of the esophagus and gastric cardia. Cancer Causes Control 1993; 4: 123. [ Links ]
7. Howson CP, Hiyama T, Wynder EL. The decline in gastric cancer: epidemiology of an unplanned triumph. Epidemiol Rev 1986; 8: 1-27. [ Links ]
8. Coleman M, Babb P, Damiecki P, Honjo S, Jons J, Knerer G. Cancer survival trends in England and Wales, 1971-1995: deprivation and NHS Region. Studies in Medical and Population Subjets nº 61. National statistics, London, 1999. [ Links ]
9. Stadtländer CT, Waterbor JW. Molecular epidemiology, pathogenesis and prevention of gastric Cancer. Carcinogenesis 1999; 20: 2195-208. [ Links ]
10. Hemminki K, Jiang Y. Familial and second esophageal cancers: a nation-wide epidemiologic study from Sweden. Int J Cancer 2002; 98: 106-9. [ Links ]
11. Kelley JR, Duggan JM. Gastric cancer epidemiology and risk factors. J Clin Epidemiol 2003; 56: 1-9. [ Links ]
12. Mowat C, Carswell A, Wirz A, McColl KE. Omeprazole and dietary nitrate independently affect levels of vitamin C and nitrite in gastric juice. Gastroenterology. 1999; 116: 813-22. [ Links ]
13. Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984; 16: 1311-4. [ Links ]
14. Bodger K, Crabtree JE. Helicobacter pylori and gastric inflammation. Br Med Bull 1998; 54: 139-50. [ Links ]
15. Forman D. Helicobacter pylori infection and Cancer. Br Med Bull 1998; 54: 71-8. [ Links ]
16. IARC Monogr Eval Carcinog Risks Hum. Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon 1994; 61: 1-241. [ Links ]
17. Farthing MJ. Helicobacter pylori infection: an overview. Br Med Bull 1998; 54: 1-6. [ Links ]
18. Cover TL, Blaser M J. Helicobacter pylori: a bacterial cause of gastritis, peptic ulcer disease, and gastric Cancer. Am Soc Microbiol News 1995; 61: 21-6. [ Links ]
19. Asghar RJ, Parsonnet J. Helicobacter pylori and risk for gastric adenocarcinoma. Semin Gastrointest Dis 2001; 12: 203-8. [ Links ]
20. Reed PI, Hill MJ, Johnston BJ. Gastric cancer and Helicobacter pylori. Lancet 1993; 16: 987-8. [ Links ]
21. Kikuchi S. Epidemiology of Helicobacter pylori and gastric cancer. Gastric Cancer 2002; 5: 6-15. [ Links ]
22. Fontana V, Decensi A, Orengo MA, Parodi S, Torrisi R, Puntoni R. Socioeconomic status and survival of gastric cancer patients. Eur J Cancer 1998; 34: 537-42. [ Links ]
23. Solcia E, Fiocca R, Luinetti O, Villani L, Padovan L, Calistri D, et al. Intestinal and diffuse gastric cancers arise in a different background of Helicobacter pylori gastritis through different gene involvement. Am J Surg Pathol 1996; 20 (Supl. 1): S8-22. [ Links ]
24. Watanabe S, Tsugane S, Yamaguchi N. Etiology. In: Sugimura T, Sasako M, eds. Gastric Cancer Oxford University Press, 1997. p. 33-51. [ Links ]
25. Tokunaga M, Land CE, Uemura Y, Tokudome T, Tanaka S, Sato E. Epstein-Barr virus in gastric carcinoma. Am J Pathol 1993; 143: 1250-4. [ Links ]
26. Fukayama M, Hayashi Y, Iwasaki Y, Chong J, Ooba T, Takizawa T, et al. Epstein-Barr virus-associated gastric carcinoma and Epstein-Barr virus infection of the stomach. Lab Invest 1994; 71: 73-81. [ Links ]
27. Shibata D, Weiss LM. Epstein-Barr virus-associated gastric adenocarcinoma. Am J Pathol 1992; 140: 769-74. [ Links ]
28. Ojima H, Fukuda T, Nakajima T, Nagamachi Y. Infrequent overexpression of p53 protein in Epstein-Barr virus-associated gastric carcinomas (abstract). Jpn J Cancer Res 1997; 88: 262-6. [ Links ]
29. Nomura A, Yamakawa H, Ishidate T, Kamiyama S, Masuda H, Stemmermann GN, et al. Intestinal metaplasia in Japan: association with diet. J Natl Cancer Inst 1982; 68: 401-5. [ Links ]
30. Kato I, Tominaga S, Ito Y, Kobayashi S, Yoshii Y, Matsuura A, et al. Atrophic gastritis and stomach cancer risk: cross-sectional analyses. Jpn J Cancer Res 1992; 83: 1041-6. [ Links ]
31. Gordon L. Tumors of the Stomach. En: Sleisenger, Fordtran's, eds. Gastrointestinal and Liver Disease. Philadelphia: Saunders. 2002. p. 733-49. [ Links ]
32. Correa P, Chen VW. Gastric cancer. Cancer Surv 1994; 19-20: 55-76. [ Links ]
33. Hsing AW, Hansson LE, McLaughlin JK, et al. Pernicious anemia and subsequent cancer: a population based cohort study. Cancer 1993; 71: 745-50. [ Links ]
34. Werner M, Becker KF, Keller G, Hofler H. Gastric adenocarcinoma: pathomorphology and molecular pathology. J Cancer Res Clin Oncol 2001; 127: 207-16. [ Links ]
35. Hsu CT, Ito M, Kawase Y, Sekine I, Ohmagari T, Hashimoto S. Early gastric cancer arising from localized Menetrier's disease (abstract). Gastroenterol Jpn 1991; 26: 213-7. [ Links ]
36. Stolte M. Clinical consequences of the endoscopic diagnosis of gastric polyps. Endoscopy 1995 ; 27: 32-7. [ Links ]
37. Parsonnet J, Friedman GD, Vandersteen DP, Change Y, Vogelman JH, Orentreich N, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 1991; 325: 1127-31. [ Links ]
38. Blot VJ, Devesa SS, Kneller RW. Rising incidence of adenocarcinoma of the esophagus and gastric cardia. JAMA 1991: 265; 1287-9. [ Links ]
39. Clark GW, Smyrk TC, Buriles P. Is Barrett's metaplasia the source of adenocarcinomas of the cardia? Arch Surg 1994; 129: 609-14. [ Links ]
40. Tahara E. Molecular biology of gastric Cancer. World J Surg 1995; 19: 484-90. [ Links ]
41. Fuchs CS, Mayer RJ. Gastric carcinoma. N Engl J Med 1995; 333: 32-41. [ Links ]
42. Yasui W, Oue N, Kuniyasu H, Ito R, Tahara E, Yokozaki H. Molecular diagnosis of gastric cancer: present and future. Gastric Cancer 2001; 4: 113-21. [ Links ]
43. Ochia, A, Hirohashi S. Multiple genetic alterations in gastric Cancer In: Sugimura T, Sasako M, eds. Gastric Cancer Oxford University Press. 1997. p. 87-99. [ Links ]
44. Wright PA, Williams GT. Molecular biology and gastric carcinoma. Gut 1993; 34: 145-7. [ Links ]
45. Tahara E. Molecular mechanism of stomach carcinogenesis. J Cancer Res Clin Oncol 1993; 119: 265-72. [ Links ]
46. Correa P, Shiao Y H. Phenotypic and genotypic events in gastric carcinogenesis. Cancer Res 1994; 54 (Supl. 7): 1941s-3s. [ Links ]
47. Tahara E, Semba S, Tahara H. Molecular biological observations in gastric Cancer. Semin Oncol 1996; 23: 307-35. [ Links ]
48. Solcia E, Fiocca R, Luinetti O, Villani L, Padovan L, Calistri D, et al. Intestinal and diffuse gastric cancers arise in a different background of Helicobacter pylori gastritis through different gene involvement. Am J Surg Pathol 1996; 20: S8-22. [ Links ]
49. Cho J-H, Noguchi M, Ochiai A, Hirohashi. Loss of heterozygosity of multiple tumor suppressor genes in human gastric cancers by polymerase chain reaction. Laboratory Investigation 1996; 74: 835-41. [ Links ]
50. Uchino S, Tsuda H, Noguchi M, Yokota J, Terada M, Saito T, et al. Frequent loss of heterozygosity at the DCC locus in gastric Cancer. Cancer Res 1992; 52: 3099-102. [ Links ]
51. Correa P. Helicobacter pylori and gastric carcinogenesis. Am J Surg Pathol 1995; 19 (Supl. 1): S37-43. [ Links ]
52. Baffa R, Veronese ML, Santoro R, Mandes B, Palazzo JP, Rugge M, et al. Loss of FHIT expression in gastric carcinoma. Cancer Res 1998; 58: 4708-14. [ Links ]
53. Lauren P. The two histological main types of gastric carcinoma; diffuse and so-called intestinal-type carcinoma. Acta Pathol Microbiol Scand 1965; 64: 31-49. [ Links ]
54. O'Connor F, Buckley M, O'Morain C. Helicobacter pylori: the cancer link. J R Soc Med 1996; 88: 674-8. [ Links ]
55. Filipe MI. Natural history of precursor lesions to gastric carcinoma: growth factors and oncogenes in the metaplasia-dysplasia-carcinoma sequence. Eur J Cancer Prev 1994; 3 (Supl. 2): 19-23. [ Links ]
56. Stemmermann G, Heffelfinger SC, Noffsinger A, Hui YZ, Miller MA, Fenoglio-Preiser CM. The molecular biology of esophageal and gastric cancer and their precursors: oncogenes, tumor suppressor genes, and growth factors. Human Pathol 1994; 25: 968-81. [ Links ]
57. Rokkas T, Filipe MI, Sladen GE. Detection of an increased incidence of early gastric cancer in patients with intestinal metaplasia type III who are closely followed up. Gut 1991; 32: 1110-3. [ Links ]
58. Werner M, Becker KF, Keller G, Hofler H. Gastric adenocarcinoma: pathomorphology and molecular pathology. J Cancer Res Clin Oncol 2001; 127: 207-16. [ Links ]
59. Rugge M, Leandro G, Farinati F, Di Mario F, Sonego F, Cassaro M, et al. Gastric epithelial dysplasia. How clinicopathologic background relates to management. Cancer 1995; 76: 76-82. [ Links ]
60. Rugge M, Farinati F, Baffa R, Sonego F, Di Mario F, Leandro G, et al. Gastric epithelial dysplasia in the natural history of gastric cancer: a multicenter prospective follow-up study. Interdisciplinary Group on Gastric Epithelial Dysplasia. Gastroenterology 1994; 107: 1288-96. [ Links ]


{kind=link}