










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.96 no.9 Madrid sep. 2004
| TRABAJOS ORIGINALES |
Diversas formas clínicas de presentación de la enfermedad celiaca dentro
de la misma familia
L. Rodrigo, S. Riestra1, D. Fuentes, S. González, A. López-Vázquez y C. López-Larrea
Servicio de Aparato Digestivo. Hospital Central de Asturias. 1Hospital Valle del Nalón. Oviedo
RESUMEN
Presentamos un estudio familiar de enfermedad celiaca, en el que se estudiaron un total de 34 miembros (8 hermanos, 23 hijos y sobrinos y 3 nietos) de un paciente adulto varón de 58 años con enfermedad celiaca (EC) complicada con afectación neurológica manifestada como mioclonías.
Encontramos 3 miembros más afectos de EC (1 hermana de 44, 1 sobrina de 39 y un sobrino de 26 años). Dos de ellos se encontraban completamente asintomáticos y todos presentaban hipertransaminasemia. Todos tenían atrofia de la mucosa duodenal (1 leve, 1 moderada y 1 importante). La afectación global familiar fue del 11,8% (4/14).
Resaltamos la importancia de realizar estudios familiares extensos ante todo nuevo diagnóstico de EC, por la elevada incidencia de agregación familiar y predominio de formas asintomáticas y señalar que el riesgo no está limitado únicamente a familiares de primer grado.
Palabras clave: Formas clínicas. Enfermedad celiaca. Estudio familiar.
INTRODUCCIÓN
La enfermedad celiaca (EC) o enteropatía por sensibilidad al gluten se caracteriza por una gran heterogeneidad en sus formas de presentación clínica (1). Los estudios de cribado de enfermedad celiaca en población general, han demostrado una elevada prevalencia de la misma (1 de cada 100-300 personas), así como una baja tasa de diagnósticos (2,3). La enfermedad celiaca no se presenta únicamente en la edad pediátrica, ya que el inicio de la misma puede retrasarse hasta edades juveniles o adultas; en nuestro medio, el 60% de pacientes con enfermedad celiaca han sido diagnosticados en edad adulta (4); algunos autores sugieren que la mayoría de estos casos se trata de formas no clásicas o atípicas de la enfermedad (5). Es por ello que, para aumentar el número de pacientes diagnosticados, debamos conocer las manifestaciones clínicas no clásicas de la enfermedad, así como realizar cribado serológico a los grupos de mayor riesgo (familiares de celiacos, diabéticos tipo I, etc.).
Los familiares de los pacientes celiacos son uno de los grupos mejor caracterizados, estando el riesgo elevado en relación con la base genética de la enfermedad (6). Múltiples estudios han demostrado una elevada prevalencia (2-18%) de enfermedad celiaca entre familiares (7-10). Esta amplia variación en los resultados, se ha puesto en relación con el método diagnóstico utilizado (serología, clínica, permeabilidad intestinal o biopsia intestinal), y con los criterios usados para diagnosticar la misma (atrofia vellositaria, formas latentes). Actualmente, el cribado serológico con la determinación de los anticuerpos antitransglutaminasa tisular (anti-TGt), constituye el método de elección para la búsqueda de nuevos casos en los grupos de riesgo.
En el presente trabajo presentamos los resultados de un amplio estudio familiar llevado a cabo a partir de un paciente con enfermedad celiaca y manifestaciones neurológicas; destacamos las formas de presentación no clásicas de la enfermedad en los familiares afectos.
CASOS CLÍNICOS
Evaluamos un total de 34 familiares (8 hermanos, 23 hijos y sobrinos y 3 sobrinos nietos) de un paciente adulto con enfermedad celiaca y síntomas neurológicos graves (caso índice). A todos los individuos se les realizó una historia clínica y exploración física completa, así como un estudio analítico que incluía: hemograma, coagulación, bioquímica general y enzimas hepáticos, niveles séricos de ferritina, ácido fólico, vitamina B-12, IgA total y TSH. Para el cribado serológico de la enfermedad celiaca determinamos los anticuerpos anti-gliadina IgA, anti-endomisio IgA y anti-transglutaminasa tisular humana IgA y estudio genético HLA-II. (fig. 1)
En los familiares en los que se detectaban alteraciones clínicas o analíticas, se realizaban los estudios específicos para filiar las mismas. Cuando existía sospecha clínica, analítica y/o serológica (anticuerpos antiendomisio y/o antitransglutaminasa tisular positivos) de EC, realizamos una endoscopia digestiva alta, con toma de múltiples biopsias duodenales.
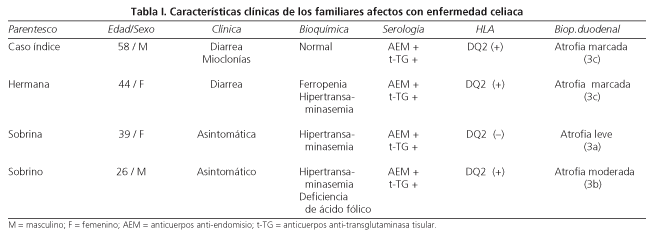
A continuación describimos las características clínicas de los 4 pacientes con enfermedad celiaca, encontrados en la misma familia, lo cual supone una afectación familiar del 11,8% (4 de 34).
Caso 1 (caso índice)
Varón de 58 años de edad, diagnosticado 3 años antes de enfermedad celiaca, tras los estudios practicados por clínica de diarrea y dolor abdominal de un año de evolución. El heterodímero HLA-DQ2 fue positivo y la biopsia intestinal mostró una atrofia vellositaria total (grado 3c de Marsh). Tras retirar el gluten de la dieta, se objetivó la desaparición de la clínica digestiva.
Hace 1,5 años, el paciente comenzó con mioclonias frecuentes e intensas. En la exploración neurológica destacaba la presencia de fasciculaciones peribucales, facies inexpresiva, bradicinesia generalizada, más marcada en hemicuerpo derecho, con fuerza conservada y mioclonías reflejas en miembro inferior derecho. Los estudios hematológicos y bioquímicos fueron normales. Los anticuerpos anti-gliadina fueron negativos, mientras que los anticuerpos anti-endomisio y antitransglutaminasa eran positivos a título bajo (1/10 y 13 U/ml, respectivamente); otros autoanticuerpos detectados eran los p-ANCA (1/80) y los anti-músculo liso (1/80). Una nueva biopsia duodenal mostró una ligera mejoría histológica, persistiendo una atrofia vellositaria parcial (grado 3a), así como un importante infiltrado inflamatorio en la lámina propia.
El estudio de células y proteínas del LCR fue normal; el cultivo del mismo fue negativo. Una RMN cerebral no mostró anomalías significativas. El registro de la actividad cerebral mediante EEG fue completamente normal. El estudio electromiográfico realizado en la pierna derecha, mostró la presencia de paroxismos punta-onda, de muy bajo voltaje, que se observaron en las regiones centrales. Se trata de un registro de mioclonías reflejas. La evaluación neuropsicológica mostró una alteración general de las funciones cognitivas, estando más preservado el lenguaje y la memoria. Todo ello sugiere una afectación predominante del hemisferio cerebral derecho, a nivel parietal, acompañado de disfunción subcortical.
El paciente fue tratado con clonazepán, piracetam, azatioprina y plasmaféresis, sin obtenerse una respuesta positiva. La afectación neurológica progresó, presentando el paciente un cuadro de desorientación, disartria y mioclonías, con imposibilidad para la deambulación. Finalmente, el paciente falleció como consecuencia de una neumonía por aspiración, dos años después del comienzo de la afectación neurológica.
Caso 2
Mujer de 44 años de edad, hermana del caso índice, con antecedentes de anemia ferropénica y diarrea ocasional. En los estudios analíticos destacaban una Hb:11,8 g/dl (VCM=71,7), AST=481 (N<31 U/l), ALT=754 (N< 31 U/l), FA=772 (N<280U/l) y GGT=134(N<32U/l). Los anticuerpos anti-VHC y antiHB-core, así como los autoanticuerpos no órgano específicos (ANA, AML, AMA y LKM-1) fueron negativos. En esta paciente se detectaron anticuerpos anti-endomisio (1/40) y anti-transglutaminasa (30 U/ml), y el heterodímero HLA-DQ2 fue positivo. El estudio de la mucosa intestinal mostró una atrofia vellositaria total (grado 3c).
Con el diagnóstico de sospecha de enfermedad celiaca, la paciente comenzó una dieta sin gluten, observándose la desaparición de la diarrea y la normalización de las cifras de hemoglobina y enzimas hepáticos a los 3 meses.
Caso 3
Mujer de 39 años, sobrina de los anteriores, asintomática, que presentaba estudios bioquímicos normales, excepto una mínima elevación de la ALT=65 (N<31 U/l) y de la GGT =146 (N<39 U/l); los anticuerpos anti-VHC, anti-HBcore, ANA, AMA, AML y LKM1 fueron negativos. Respecto al estudio serológico de enfermedad celiaca, se detectaron anticuerpos anti-endomisio (1/10) y anti-transglutaminasa tisular (14 U/ml); el heterodímero HLA-DQ2 fue negativo. La biopsia duodenal mostró una atrofia parcial de las vellosidades intestinales (grado 3a).
Tras la supresión del gluten de la dieta la paciente sigue asintomática, se han normalizado las pruebas hepáticas y se han negativizado los anticuerpos antiendomisio y antitransglutaminasa tisular.
Caso 4
Varón de 26 años, sobrino de los dos primeros y primo de la anterior, asintomático, en quien se detecta una mínima elevación de la AST=42 (N<31 U/l) y ALT=72 (N<31 U/l), así como un descenso de los niveles séricos de ácido fólico, 2 (N>3 ng/ml), sin anemia. Los anticuerpos EMA (1/320) y anti-tTG (> 200 U/ml) fueron positivos, así como el heterodímero HLA-DQ2. La biopsia duodenal mostró una atrofia vellositaria moderada (grado 3b).
Al igual que en el resto de familiares celiacos, la supresión del gluten se acompañó de la desaparición de las alteraciones analíticas y de la negativización de los anticuerpos anti-endomisio y anti-transglutaminasa tisular. Se muestra un esquema de los cuatro casos en la tabla I.
DISCUSIÓN
La primera comunicación sobre el carácter familiar de la enfermedad celiaca data de 1935 (11); actualmente se sabe que existe un 75% de concordancia para la enfermedad celiaca entre gemelos monocigotos, frente a un 11% entre gemelos dicigotos (12), lo cual es evidencia para una fuerte carga genética de la enfermedad. La asociación genética más potente es con los alelos HLA de clase II, DQA1*501 y DQB1*201, que codifican el heterodímero DQ2 (13); sin embargo, estos están presentes también en un 20% de la población general (10), por lo que parece demostrada la necesidad de otros factores genéticos o ambientales en el desarrollo de la enfermedad. Así, en nuestro grupo, hemos observado como un gen HLA de clase I, el alelo MICA 5.1, se asocia a formas no clásicas de enfermedad celiaca en sujetos con el heterodímero de alto riesgo DQ2 (14).
El primer aspecto que nos gustaría destacar del presente trabajo, es la elevada prevalencia de enfermedad celiaca en una misma familia (4 de 34; 11,8%); en alguna publicación, hasta el 2,6% de las familias estudiadas tenían tres o más miembros con enfermedad celiaca. Según el grado de relación familiar, hemos detectado casos entre hermanos y sobrinos del caso índice, lo cual apoya la necesidad de hacer estudios familiares amplios para no dejar sin diagnosticar casos dentro de una misma familia. Otro aspecto a destacar es que 2 de los 4 familiares con enfermedad celiaca estaban asintomáticos, lo cual apoya la necesidad de hacer estudio serológico a todos los familiares, no pudiendo basarnos únicamente en la presencia de síntomas para sospechar la enfermedad. El abordaje diagnóstico más eficiente, debe incluir la biopsia intestinal en todos los familiares con anticuerpos anti-transglutaminasa, pues así se ha demostrado que se diagnostican hasta el 92,3% de los casos familiares; no obstante, se ha sugerido por otros autores (15), la presencia de formas leves de la enfermedad entre familiares con serología negativa, pero con síntomas atípicos y/o alteraciones bioquímicas. Por tanto, debemos de tener en cuenta que, al igual que ocurre en la práctica clínica, ante la sospecha de enfermedad celiaca la "prueba oro' es la biopsia intestinal, independientemente del resultado de la serología.
La enfermedad celiaca se puede asociar hasta en un 8% de los casos a síntomas neurológicos, fundamentalmente ataxia cerebelosa, epilepsia y neuropatía periférica. Los pacientes con enfermedad celiaca con manifestaciones neurológicas, no suelen presentar clínica digestiva, de ahí que el diagnóstico de la sensibilidad al gluten sea generalmente tardío en estos casos. Es importante el reconocimiento de la enfermedad celiaca, puesto que la supresión del gluten precozmente tras el inicio de los síntomas neurológicos puede causar su mejoría o desaparición. La patogenia de la afectación neurológica en la enfermedad celiaca no es bien conocida, pero se ha relacionado con un origen autoinmune; en pacientes celiacos con síntomas neurológicos, se ha demostrado la presencia de anticuerpos anti-células de Purkinje y anti-neuronas del SNC en suero (16-18).
La asociación entre enfermedad celiaca y mioclonías ya ha sido comunicada previamente y se caracteriza, como en nuestro caso índice, por su inicio en la edad adulta y años después del diagnóstico de la enfermedad celiaca (19,20); en cuanto a la causa de las mioclonías se sabe que aunque su origen sea cortical, la patología generalmente se localiza a nivel del cerebelo (19). En nuestro paciente las mioclonías aparecieron varios años después del diagnóstico de enfermedad celiaca, y mientras seguía una dieta sin gluten; no obstante, la persistencia de lesión histológica en la mucosa intestinal, pudiera estar relacionada con una falta de cumplimiento del tratamiento dietético, lo cual podría explicar, en cierta manera, la aparición de las complicaciones neurológicas; aunque ciertamente, no se realizaron otras pruebas que descartaran la existencia asociada de otra enteropatía. Al igual que en otros casos de enfermedad celiaca con afectación neurológica asociada, no hubo respuesta a los tratamientos pautados, siendo además destacable en nuestro paciente, la rápida evolución del cuadro neurológico hacia una forma invalidante, que fue causa de su fallecimiento.
En tres de nuestros familiares con enfermedad hepática, la alteración bioquímica más llamativa fue una hipertransaminasemia. Al diagnóstico, aproximadamente un 30% de los celiacos tienen alteración en las pruebas hepáticas, siendo el sustrato morfológico de la misma una hepatitis reactiva inespecífica (21,22); como en nuestros casos, la instauración de una dieta sin gluten se sigue de la normalización de las alteraciones bioquímicas, por lo que no es preciso la realización de pruebas diagnósticas agresivas como la biopsia hepática. Dado que la sensibilidad al gluten es causa de un 4,4-9% de hipertransaminasemias criptogenéticas (23-25), creemos que, dentro del protocolo diagnóstico de esta alteración bioquímica, debería incluirse el estudio serológico de la enfermedad celiaca.
Como conclusión, queremos destacar en este trabajo, la necesidad de llevar a cabo estudios familiares amplios, cuando se diagnostica un caso de enfermedad celiaca, puesto que el riesgo no sólo se limita a los familiares más próximos. Por otra parte, nos gustaría llamar la atención sobre la frecuente aparición de formas atípicas o asintomáticas entre los familiares de pacientes celiacos. Con todo ello, es posible aumentar el número de pacientes diagnosticados de enfermedad celiaca, dentro de un grupo de riesgo ya bien caracterizado.
BIBLIOGRAFÍA
1. Farrell RJ, Kelly CP. Celiac sprue. N Engl J Med 2002; 346: 180-8. [ Links ]
2. Riestra S, Fernández E, Rodrigo L, García S, Ocio G. Prevalence of coeliac disease in the general population of Northern Spain. Strategies of serological screening. Scand J Gastroenterol 2000; 35: 398-402. [ Links ]
3. Mäki M, Mustalahti K, Kokkonen J, Kulmala P, Haapalahti M, Karttunen T, et al. Prevalence of celiac disease among children in Finland. N Engl J Med 2003; 348: 2517-24. [ Links ]
4. Riestra S, Fernández E, Bousoño C, Rodrigo L. Enfermedad celíaca del adulto. Gastroenterol Hepatol 2001; 24: 515. [ Links ]
5. Ivarsson A, Persson LA, Juto P, Peltonen M, Suhr O, Hernell O. High prevalence of undiagnosed coeliac disease in adults: a Swedish population-based study. J Intern Med 1999; 245: 63-8. [ Links ]
6. Schuppan D. Current concepts of coeliac disease pathogenesis. Gastroenterology 2000; 119: 234-42 [ Links ]
7. Stenhammar L, Brandt A, Wagermark J. A family study of celiac disease. Acta Paediatr Scand 1982; 71: 625-8. [ Links ]
8. Mäkki M, Holm K, Lipsanen V, Hällström O, Viander M, Collin P, et al. Serological markers and HLA genes among healthy first-degree relatives of patients with celiac disease. Lancet 1991; 338: 1350-3. [ Links ]
9. Bonamico M, Mariani P, Mazzilli MC, Triglione P, Lionetti P, Ferrante P, et al. Frequency and clinical pattern of celiac disease among sibling of celiac children. J Pediatr Gastroenterol Nutr 1996; 23: 159-63. [ Links ]
10. Farré C, Humbert P, Vilar P, Varea V, Aldeguer X, Carnicer J, et al. Serological markers and HLA-DQ2 haplotype among first-degree relatives of celiac patients. Dig Dis Sci 1999; 44: 2344-9. [ Links ]
11. Thaysen TEH. Ten cases of idiopathic steatorrhea. Q J Med 1935; 4: 359-65. [ Links ]
12. Greco L, Romino R, Coto I, Cosmo ND, Percopo S, Maglio M, et al. The first large population based twin study of coeliac disease. Gut 2002; 50: 624-8. [ Links ]
13. Sollid LM, Thorsby E. HLA susceptibility genes in celiac disease: genetic mapping and role in pathogenesis. Gastroenterology 1993; 105: 910-22. [ Links ]
14. López-Vázquez A, Rodrigo L, Fuentes D, Riestra S, Bousoño C, García-Fernández S, et al. MHC class I chain related gene A (MICA) modulates the development of coeliac disease in patients with the high risk heterodimer DQA1*501/DQB1*201. Gut 2002; 50: 336-40. [ Links ]
15. Rostami K, Mulder CJJ, van Overbeek FM, Kerckhaert J, Meijer JWR, von Blomberg MBE, et al. Should relatives of coeliacs with mild clinical complaints undergo a small-bowel biopsy despite negative serology? Eur J Gastroenterol Hepatol 2000; 12: 51-5. [ Links ]
16. Volta U, De Giorgio R, Petrolini N, Stanghellini V, Barbara G, Granito A, et al. Clinical findings and anti-neuronal antibodies in coeliac disease with neurological disorders. Scand J Gastroenterol 2002; 37: 1276-81. [ Links ]
17. Hadjivassiliou M, Gibson A, Davies-Jones GAB, Lobo AJ, Stephenson AJ, Milford-Ward A. Does cryptic gluten sensitivity play a part in neurological illness? Lancet 1996; 347: 369-71. [ Links ]
18. Hadjivassiliou M, Boscolo S, Davies-Jones GAB, Grünewald RA, Not T, Sanders DS, et al. The humoral response in the pathogenesis of gluten ataxia. Neurology 2002; 58: 1221-6. [ Links ]
19. Bhatia KP, Browm P, Gregory R, Lennox GG, Manji H, Thompson PD, et al. Progressive myoclonic ataxia associated with celiac disease. The myoclonus is of cortical origin, but the pathology is in the cerebellum. Brain 1995; 118: 1087-93. [ Links ]
20. Mumford CJ, Fletcher NA, Ironside JW, Warlow CP. Progressive ataxia, focal seizures, and malabsorption in a 41 year old woman. J Neurol Neurosurg Psychiatry 1996; 60: 225-30. [ Links ]
21. S Riestra, E Fernández, L Rodrigo. Afectación hepática en la enfermedad celíaca. Rev Esp Enferm Dig 1999; 91: 846-52. [ Links ]
22. Jacobsen MB, Fausa O, Elijo K, Schrumpf E. Hepatic lesions in adult coeliac disease. Scand J Gastroenterol 1990; 25: 656-62. [ Links ]
23. Volta U, de Franceschi L, Lari F, Molinaro N, Zoli M, Bianchi FB. Coeliac disease hidden by cryptogenic hypertransaminasaemia. Lancet 1998; 352: 26-9. [ Links ]
24. Bardella MT, Vecchi M, Conte D, Del Ninno E, Fraquelli M, Pacchetti S, et al. Chronic unexplained hypertransaminasaemia may be caused by occult celiac disease. Hepatology 1999; 29: 654-7. [ Links ]
25. Vivas S, Ruiz de Morales JM, Martínez J, González MC, Martín S, Martín J, et al. Human recombinant anti-transglutaminase antibody testing is useful in the diagnosis of silent coeliac disease in a selected group of at-risk patients. Eur J Gastroenterol Hepatol 2003; 15: 479-83. [ Links ]

