










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.101 no.3 Madrid mar. 2009
Tumores neuroendocrinos: fascinación e infrecuencia
Neuroendocrine tumors - fascination and infrequency
M. J. Varas Lorenzo
Centro Médico Teknon. Barcelona
Dirección para correspondencia
RESUMEN
Se efectúa una revisión y puesta al día, basándose en citas bibliográficas de los últimos quince años, de los tumores neuroendocrinos gastroenteropancreáticos, que tanta fascinación han provocado en el estamento médico por su infrecuencia, síndromes hormonales y supervivencia elevada.
Palabras clave: Tumores neuroendocrinos. Tumores neuroendocrinos pancreáticos. Tumores endocrinos gastroenteropancreáticos. Carcinoides.
ABSTRACT
In this article, I review and update of gastro-entero-pancreatic neuroendocrine tumors, which so much fascination have risen among healthcare providers on grounds of their infrequency, hormonal syndromes, and high survival rate, is performed based on references from the past fifteen years.
Key words: Neuroendocrine tumors. Neuroendocrine pancreatic tumors. Gastro-entero-pancreatic endocrine tumors. Carcinoid tumors.
Definición
Los tumores neuroendocrinos (TNE) o tumores endocrinos gastroenteropancreáticos (TEGEP) (2% de todos los tumores del tubo digestivo) se originan en los tejidos derivados de la cresta neural, el neuroectodermo y el endodermo, el 60-70% son carcinoides y el 20-40% están situados en el páncreas (TNEP) (Tablas I y II). Los tumores endocrinos del tracto digestivo que no son carcinoides pueden ser gastrinomas, enteroglucagonomas, somatostatinomas, o tumores neuroendocrinos no funcionantes. Muchos de los datos que aquí se manejan están tomados de anteriores revisiones (1-5).
Incidencia
En necropsias, el 0,5-1,5% y hasta el 5% son TNEP (1,2), muchos silentes. Representan el 1-3% de todas las neoplasias del páncreas.
En Europa, en el único estudio europeo de base comunitaria, la incidencia es de menos de 1 caso por 100.000/año, concretamente de 0,32 por 100.000/año de carcinoides, y 0,2 por 100.000/año (2 por millón/año) en el páncreas (6). El insulinoma y el gastrinoma, que son los más frecuentes, tendrían una incidencia de 1 caso nuevo por millón de habitantes y año. Los glucagonomas y los somatostatinomas son los menos frecuentes (1 por 20 y 40 millones/año, respectivamente) (Tabla II).
Los carcinoides se presentan el doble en pacientes afroamericanos que en pacientes blancos (7).
En EE. UU. (8), casi 5 por millón/año para los TNEP, 1,8 en mujeres y 2,6 en varones, aunque la incidencia aumenta, y pudiera llegar a 10 por millón de habitantes y año, con una mayor frecuencia entre los 40 y 60 años, y en mujeres.
En nuestro país, la incidencia de los TNEP pudiera ser de 0,08 por 100.000 habitantes/año, o 1 caso nuevo cada dos años en Hospitalet de Llobregat (Barcelona).
Genética
Pueden ser tumores esporádicos, sobre todo los carcinoides, o no esporádicos asociados a síndromes con cromosoma autosómico dominante (2), cromosoma locus 11q13 en la NEM-1, que incluyen la neoplasia endocrina múltiple (NEM) tipo 1 y 2 (a y b), la enfermedad de von Hippel-Lindau (VHL), la neurofibromatosis de von Re-cklinghausen o la esclerosis tuberosa (5) (Tabla III).

El NEM 2 resulta de la mutación del protooncogén RET.
El riesgo de desarrollar carcinoides también se explica por factores genéticos; en efecto, existe un aumento de los TNE gástricos en mujeres con diabetes y antecedentes familiares de cáncer (9).
Tumores funcionantes y no funcionantes (TNENF)
Los TNENF se diagnostican durante la ecografía de rutina (Fig. 1), el TC para el estudio de síntomas abdominales inexplicables, o en fases tardías de la enfermedad (el 70% son mayores de 5 cm).
Su frecuencia es del 20-58%, y hasta el 90% cuando se localizan en el páncreas (10-12).
La mayoría (más del 70%) no son verdaderamente funcionantes, ya que secretan sustancias como el PP, otros péptidos (PYY, Ghrelina, etc.), ENE, CgA, HCG, aunque ninguna de estas sustancias causan síntomas específicos.
Cuando son funcionantes (60%) o secretantes pueden dar lugar a secreción hormonal múltiple (SHM) o estar asociados a NEM, con síndromes bien definidos.
Excepto el insulinoma (menos del 10%), muchos TNE son malignos (50-60% y porcentajes más altos, sobre todo los TNEPNF) con metástasis ganglionares, hepáticas (las más frecuentes), óseas (12%), pulmonares (4%) y cerebrales (Tabla II).
La supervivencia a los 5 años de todos los TNE no sobrepasa el 67%.
Recientemente la OMS ha creado una clasificación para los TNE: tumor bien diferenciado, carcinoma bien diferenciado y carcinoma poco diferenciado, funcionalidad de la histología, tamaño e índice de proliferación, funcionalidad si bien anteriormente se había propuesto una clasificación TNM (tumor, nódulo, metástasis) (5).
Sospecha clínica
Cuando los TNEP segregan péptidos y hormonas de forma inapropiada (TNEPF) dan lugar a síndromes clínicos específicos con síntomas guías (13-15). Así la aparición de hipoglucemia aguda y/o crónica (síntoma guía) debe hacer sospechar un hiperinsulinismo por insulinoma pancreático (Tabla II).
La aparición de dolor abdominal, vómitos y diarrea, en un paciente ulceroso sobre todo si es Helicobacter pylori negativo, debe alertar sobre la existencia de un síndrome de Zollinger-Ellison (SZE) por gastrinoma pancreático o extrapancreático (14).
El síndrome del glucagonoma se caracteriza por eritema necrolítico migratorio (ENM) (52%) y diabetes (22%) (16); otros como el somatostatinoma y el corticotrofinoma también se asocian a hiperglicemia.
El síndrome de Verner-Morrison (SVM) por vipoma asocia diarrea secretora a hipopotasemia (70-100%). Muchos TNEP producen diarrea crónica, pero sólo el vipoma y neurotensinoma producen diarrea hipopotasémica.
El síndrome clínico "inhibitorio" del somatostatinoma asocia diabetes a patología vesicular, diarrea e hipoclorhidria. El somatostatinoma puede localizarse en el duodeno o en el páncreas, con una malignidad mayor del 43% (17).
El flushing es bastante típico del síndrome carcinoide (SC) que aparece en el 10% de los tumores carcinoides (Tabla IV).

La sospecha clínica debe obligar al médico a solicitar una batería hormonal (no sólo la hormona en cuestión sino que además hay que descartar SHM y NEM); no obstante, en ocasiones los tumores no son funcionantes o los niveles hormonales se encuentran dentro de los límites de la normalidad, por lo que se debe recurrir a tests de estimulación (14,15,18).
Diagnóstico bioquímico
El polipéptido pancreático (PP), la enolasa neuronal específica (ENE), cromograninas (A, B y C), sinaptofisina (P38), 7B2 y HCG, son marcadores inespecíficos, que tienen especial relevancia en el diagnóstico de los TEPNF.
La CgA y la ENE son excelentes marcadores de TEGEP, mientras que Ki 67 (mayor del 2%) o MIB-1 es un marcador pronóstico de malignidad (Tabla V). Probablemente la CgA es más específica y la ENE posee mayor sensibilidad (83-100%).

La CgA posee una sensibilidad del 60-100% en pacientes con metástasis, pero de menos del 50% en pacientes con tumores localizados (15).
En los TNEPF y carcinoides, la insulina, glucagón, somatostatina, gastrina, VIP, PP, serotonina (y su metabolito 5-HIAA urinario), etc., se dosifican por RIA.
Esta batería hormonal por RIA es importante no sólo para el diagnóstico bioquímico si no también para el seguimiento, para monitorizar la respuesta a la quimioterapia y a la cirugía (14,15).
Es necesario también efectuar investigaciones sobre la PTH, calcio, calcitonina, prolactina, etc.
Diagnóstico y localización por la imagen
Los TNE del tracto digestivo (fundamentalmente carcinoides) son localizables por radiología y endoscopia (Fig. 2), y se pueden estadificar por ultrasonografía endoscópica (USE) que informa sobre la profundidad y la extensión del tumor y ello, a su vez, facilita la tumorectomía o polipectomía endoscópica (carcinoides de 1-2 cm, localizados en primeras capas, que no infiltran la muscular propia ni tienen adenopatías). Debe descartarse enfermedad metastásica o tumores ocultos mediante Octreoscan y PET.
Los TNEP son más difíciles de localizar; casi el 20-30% de los casos se pueden localizar mediante los métodos de diagnóstico por la imagen (ecografía, TCH, RM con gadolinium, gammagrafía, Octreoscan, etc.).
TC y RM detectan del 30-94% de los casos, y Octreoscan del 80-90% (a excepción del insulinoma: 50%), mientras que la USE detecta del 79-100% (5).
Efectivamente la USE pancreática logra visualizar tumores muy pequeños de hasta 3 mm, con una sensibilidad superior al 85% cuando están localizados en la cabeza y cuerpo intrapancreático; incluso, en un estudio que analiza una muestra más amplia alcanzan hasta el 93% (19).
En la revisión de la literatura (20-22), la sensibilidad media es alta, del 88% y la especificidad del 85%, por lo tanto quedarían todavía alrededor de un 5-10% de los tumores sin localizar preoperatoriamente.
En nuestra experiencia acumulada con 50 enfermos, 35 de los cuales tenían 34 tumores operados (14 en el páncreas y 20 en el tracto digestivo, 15 carcinoides), la precisión de la USE fue del 82,5%, la sensibilidad del 88% (94% para los carcinoides, 90% para los del tracto digestivo, 86% para los pancreáticos) y el VPP del 91%. En el páncreas se detectaron 3 tumores menores de 10 mm, 4 menores de 15 mm, todos insulinomas, y 1 tumor quístico de 4 mm (2%) que era un gastrinoma funcionante.
La evolución a lo largo de 10 años indica un aumento paulatino de la precisión y sensibilidad diagnóstica, fruto de la experiencia (22).
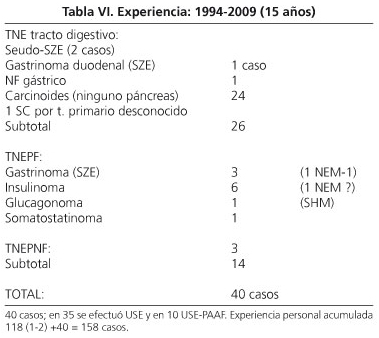
En 10 casos (8 operados) se practicó USE-PAAF, con una precisión del 75%, una sensibilidad y VPP del 83%, y una especificidad y VPN del 50%, ya que hubo un falso positivo (Tabla VI y Fig. 3).
Se ha propuesto un algoritmo para la localización y estadificación basado en la práctica de un TC helicoidal de última generación y USE radial y sectorial si es necesario obtener material histológico con PAAF, con precisiones y sensibilidades muy variadas en la literatura, pero que de media superan el 80%, y al 90% en la serie con más casuística con PAAF (23).
En algunos casos, 1,5-4% (24,25), sobre todo cuando los TNEP son muy pequeños, la mayoría no funcionantes (24-27), pueden presentar un patrón quístico, de tal manera que la USE-PAAF puede ofrecer muy buenos resultados (26,27), casi del 100%.
Se estima que la prevalencia de los TNEP puede ser más alta, del 9,5% (26), aunque nuestra experiencia (2%) es similar a la encontrada en la literatura (25).
En la actualidad probablemente la mejor estrategia sea practicar TCH más USE con PAAF opcional. El Octreoscan podrá ser remplazado en el futuro por el PET con octreotido, que tiene una sensibilidad, especificidad y precisión mayor del 90%.
De todas formas, en el futuro, es muy pobable que todos estos planteamientos habrán de cambiar con las nuevas tecnologías, tales como la USI y la USE con color o contrastes tipo Sonovue (56), la sonoelastografía, la ultrasonografía intraductal con minisondas, el PET, etc.
Posibilidades terapéuticas
1. Cirugía. El tratamiento quirúrgico clásico o con laparoscopia, con exéresis total representa la única alternativa curativa actual, después de estabilizar al paciente (fluidos, diazóxido, IBP, insulina, según tumor, etc.).
La polipectomía endoscópica está indicada en los carcinoides del tracto digestivo, menores de 1-2 cm, localizados en primeras capas, sin afectar a la muscular propia y en ausencia de adenopatías (22).
2. Somatostatina y análogos (octreotida, lanreótida, pasireótida). Con acción inhibitoria de la secreción hormonal. Se pueden administrar por vía subcutánea o intramuscular cada día, o cada 7-14-30 días, a dosis que oscilan entre 100 y 1.500 mcg/día en dos o tres veces subcutáneamente. En el caso de la administración intramuscular la dosis será de 20-30 mg cada 7-14-28 días según respuesta, inclusive 20 mg cada mes i.m. No producen regresión (3-14%) pero sí estabilización tumoral del 36-70%, y disminución de los paramétros clínicos (más del 50%) y bioquímicos (43%) (28).
3. Interferones. El interferón leucocitario humano y el interferón recombinante humano alfa-2B se han administrado por vía subcutánea, este último a dosis de 2 a 6 millones UI/día, durante varias semanas, con respuesta tumoral del 11% y respuesta bioquímica de 44% de media (29,30). Según un reciente estudio con interferón alfa Pegylato la enfermedad se estabilizaría en el 75% de los casos (31) con control sintomático.
4. Quimioterapia. Con estreptozotocina (1 g i.v./m2/día en ciclos de 5-7 días cada 6 semanas), estreptozotocina más 5-FU, o doxorubicina, producen remisiones que llegan hasta el 60%. También se puede utilizar una combinación de cisplatino y etopósido en TNE pobremente diferenciados.
Se usan temozolomida y talidomida oral en pacientes con metástasis avanzadas (32).
5. Radioterapia y otras. Radioterapia paliativa, In111-DTPA octreotido para tumores no resecables que tengan Octreoscan positivo, con parciales remisiones y estabilización del tamaño tumoral en más del 60% de los casos.
Los TNE pueden producir múltiples factores de crecimiento, factor de crecimiento vascular endotelial, factor de crecimiento plaquetar, factor de crecimiento similar a la insulina, factor de crecimiento de fibroblastos y factor de crecimiento transformante, factor de crecimiento epidérmico, así como receptores para estos factores. Nuevas moléculas dirigidas hacia estos factores o sus receptores, anticuerpos monoclonales, inhibidores de la mTOR, inhibidores de tirosinquinasa como el sunitinib, que no alcanzan el 20% de las respuestas (5), aunque en uno de los últimos trabajos publicados (33) sobre 107 pacientes el 44% de los carcinoides y el 62% TNEP tuvieron al menos alguna regresión y estabilización tumoral.
6. Combinaciones de las anteriores. Por ejemplo análogos de la somatostatina con alfa-interferón (30) o quimioterapia.
7. En las metástasis hepáticas. Quimioembolización, etanolización, criocirugía y ablación por radiofrecuencia: y en último término se puede plantear el trasplante hepático en pacientes jóvenes con metástasis hepáticas y TNEP primario extirpado (15).
Pronóstico y supervivencia
El pronóstico de los carcinoides es muy malo cuando aparece síndrome carcinoide (10% de los casos) ya que es manifestación de una fase avanzada de la enfermedad (metástasis hepáticas).
El pronóstico de los TNEP depende de la funcionalidad, de la existencia o no de NEM, del tamaño y localización del tumor primitivo, y de la presencia de metástasis (extensión del tumor) (14).
En la estadística del H. General de Massachussets (168 TNEP) el 76% fueron benignos y el 26% malignos con metástasis hepáticas. La supervivencia actuarial fue del 77 y 62% a los 5-10 años (10).
En los TNEP benignos la supervivencia a los 5 años fue del 92 frente al 50% de los malignos (11).
Hoy en día, la principal preocupación del especialista en relación con el SZE es la aparición de metástasis hepáticas que representan el más importante de los factores de mal pronóstico.
Cuando se opera con intención curativa, el gastrinoma tiene una supervivencia a los 15 años del 98 frente al 74% cuando el paciente no es quirúrgico (34). El 29% de los enfermos no operados desarrollaron metástasis hepáticas.
En la estadística del H. Princesa Margaret (193 TNE en 10 años: 72% carcinoides y 21% TNEP) la supervivencia a los 5 años fue del 58%, y en el análisis multivariante, la edad, localización primaria tumoral, y la cirugía con intento curativo fueron factores independientes predictores de supervivencia. De estos pacientes cuando hubo un intento de cirugía curativa la supervivencia a 5 años subió al 86% (35).
En la estadística de la Clínica Mayo (1.483 TNE en 27 años, con un alto porcentaje de no funcionantes: 91%) el estado avanzado y la edad fueron predictores de peor supervivencia, obteniéndose mejores resultados en los funcionantes que en los no funcionantes (8).
Cuando se operan pacientes con NEM-1 poseen una similar supervivencia a los 7-10-15 años, pero tienen una alta recurrencia, y sólo un 4,5% están libres de tumores a los 10 años (36).
La supervivencia de los TNE sin metástasis hepáticas fue del 95-90-83% a los 5-10-15 años respectivamente; pero incluso con enfermedad maligna, la supervivencia a 5 años puede ser alta del 77-95%, cuando se efectúa un tratamiento agresivo con resección primaria del tumor y terapia adyuvante, comparado con el 36% a los 5 años de otros estudios (4).
Diferentes aspectos se relacionan con un pronóstico malo, tamaño tumoral mayor de 2 cm, presencia de invasión vascular o perineural, infiltración de la cápsula pancreática, la diferenciación histológica (pobremente diferenciado), número de mitosis, la atipia celular, el Ki 67 alto (mayor del 30%), y la presencia de metástasis hepáticas o linfáticas.
En 70 pacientes operados por TNEP (23% insulinomas, 71% no funcionantes y 53% malignos) la supervivencia actuarial a los 5 años fue del 77%. Los ganglios, la invasión perineural y las metástasis hepáticas no tuvieron impacto en la supervivencia, solamente la invasión linfovascular (37).
Autores alemanes (38,39) con una estadística de casi 400 TNE, 70% con metástasis en el diagnóstico inicial, creen que el tamaño tumoral menor de 2,5 cm, la intervención quirúrgica, la ausencia de metástasis y de síntomas, la CgA y el índice Ki 67 bajos, son los mejores marcadores de supervivencia.
El grupo sueco (40) estudia los factores pronósticos sobre 324 pacientes con TNEP, con una supervivencia a los 5 y 10 años, del 64 y 44%, respectivamente. En el análisis univariante, el estadio, la cirugía radical, el ser no funcionante, el Ki 67 y la CgA elevados, el tamaño tumoral y ser esporádico (no familiar) son factores pronósticos significativos; mientras que en el análisis multivariante sólo lo fueron el estadio, la cirugía radical y el Ki 67 mayor de 2%.
Autores italianos (41) estudian 180 casos de TNEPNF, con supervivencias a los 5, 10 y 15 años del 67, 49 y 33% respectivamente, confirmando que las metástasis, la pobre diferenciación del tumor, el Ki 67 y la pérdida de peso son factores con valor pronóstico (Tabla V).
Autores americanos (42) sobre un programa de registro de 35.825 casos concluyen que en el análisis multivariante los factores predictivos son la diferenciación tumoral, el estadio, la localización primaria tumoral, el grado histólogico, sexo, raza, edad y año de diagnóstico; confirmando un incremento de su incidencia, pasando de 1 por 100.000 en el año 1973, a 5,25 por 100.000 en el año 2004.
Estos datos coinciden al comparar la incidencia (1993-2004) en EE. UU. y en Noruega, 4,44 versus 3,24 en pacientes caucasianos, aunque es más alta en pacientes afroamericanos. Siempre el tumor más frecuente fue el broncopulmonar (32%). La supervivencia a los 5 años de todos los TNE fue del 55% (43).
Con respecto a los TNE broncopulmonares, la mayoría son carcinoides, poco funcionantes (-5%) con síndrome de Cushing (SC), acromegalia y SIADH. Su incidencia en 2003 fue de 1,57 por 100.000, aumentando. La supervivencia a los 5 años fue del 88% para los tumores carcinoides típicos con bajo crecimiento (44).
Anteriormente se consideraba que los TEGEP superaban a los broncopulmonares, si bien en uno de los últimos registros sobre 1.000 casos en Brasil, el 72% estaban en el tórax, el 20% eran TEGEP, y un 3,6% en lugar desconocido (45).
Resumen y conclusiones
Los tumores neuroendocrinos (TNE) o gastroenteropancreáticos (TEGEP) pueden ser carcinoides en el 70% y pancreáticos en el 20% de los casos.
Su incidencia es de más de 5 casos por millón de habitantes y año, y está aumentando.
Pueden ser esporádicos o ligados a mutación genética.
Pueden ser funcionantes (50%) o no funcionantes; benignos o malignos en función de la existencia de enfermedad metástasica (50-60%).
Su sospecha clínica debe alertar al médico a solicitar una batería hormonal, con marcadores inespecíficos como la CgA y la ENE, o marcadores específicos (insulina, gastrina, VIP, serotonina -5-HIAA-, etc.).
Actualmente, el algoritmo diagnóstico para los carcinoides del tracto digestivo incluye la endoscopia con biopsia, el TACH y el Octreoscan vs. PET.
Para los TNEP y la NEM, la mejor estrategia parece ser la práctica de un TACH más USE radial o sectorial con PAAF opcional (Fig. 3), seguido de Octreoscan vs. PET.
El PET con 68Ga-DOTA-Tyr3-octreotrido tiene una sensibilidad del 97%.
Después de estabilizar al paciente por la secreción hormonal inapropiada (fluidos, diazóxido, IBP, insulina, etc.), si no hay metástasis, debe intentarse la cirugía con intención curativa, y si hay metástasis añadirle terapia adyuvante.
En los casos con enfermedad avanzada se puede efectuar tratamiento con análogos de la somatostatina, interferones, ambos, o incluso quimioterapia o radioterapia con derivados del octreotido, cuando el Octreoscan es positivo.
El pronóstico y la supervivencia, fundamentalmente, están en función de la posibilidad de realizar cirugía con intención curativa y de la ausencia de enfermedad metastásica avanzada, y probablemente de la invasión linfovascular (37).
Otros factores importantes además serían el tamaño del tumor primario pancreático mayor de 2,5 cm, la CgA y el Ki 67 elevado (38,39,40); y en los TNEPNF las metástasis, el Ki 67 y la pérdida de peso (41).
En los carcinoides (Fig. 4) del tracto digestivo menores de 1 cm, la polipectomía endoscópica es satisfactoria y ha sustituido a la cirugía clásica, si el tumor no infiltra la muscular propia y no ha desarrollado metástasis (46). En estos casos y en los carcinoides apendiculares la supervivencia a los 5 años es mayor del 95%, mientras que desciende al 25 y 65% si existen metástasis hepáticas o ganglionares (47-49).
Pese a que en los últimos años se han producido avances en el diagnóstico y tratamiento de los TNEP (48-50), no está claro que haya mejorado la supervivencia. En una reciente revisión (51) no se registraron cambios en los índices de supervivencia durante 30 años, para todos los TEGEP (carcinoides y TNEP) (52).
Recientes trabajos defienden la resección quirúrgica de los tumores pancreáticos (53), aunque en los pacientes con enfermedad metastásica la resección del tumor primario no beneficia a los carcinoides derivados del intestino medio (54).
También muy recientemente se han descrito 4 casos de TEGEP en pacientes con VIH (55).
![]() Dirección para correspondencia:
Dirección para correspondencia:
M. J. Varas Lorenzo.
Centro Médico Teknon.
Marquesa de Vilallonga, 12.
08017 Barcelona, Spain.
e-mail: varas@dr.teknon.es
Recibido: 28-12-08.
Aceptado: 08-01-09.
Bibliografía
1. Varas MJ. Endocrinología Gastroentero-pancreática. Madrid: Smar SL.; 1997. [ Links ]
2. Varas MJ. Tumores de los islotes pancreáticos. En: Vilardell F, et al., editores. Enfermedades Digestivas 2. Madrid-Barcelona: Aula Médica; 1998. p. 1516-24. [ Links ]
3. Fritscher-Raven A. Endoscopic ultrasound and neuroendocrine tumours of the pancreas. JOP 2004; 5(4): 273-81. [ Links ]
4. Mougey AM, Adler DG. Neuroendocrine tumors: review and clinical update. Hospital Physician 2007. p. 12-20. [ Links ]
5. Massironi S, Sciola V, Peracchi M, Ciafardini C, Spampatti MP, Conte D. Neuroendocrine tumors of the gastro-entero-pancreatic system. World J Gastroenterol 2008; 14(35): 5377-84. [ Links ]
6. Buchanan KD, Johnston CF, O'Hare M, Ardill JE, Shaw C, Collins JS, et al. Neuroendocrine tumors: a European view. Am J Med 1986; 81: 14-27. [ Links ]
7. Kang H, O'Connell JB, Leonardi MJ, Maggard MA, MxGory ML, Ko CY. Rare tumors of the colon and rectum: a national review. Int J Colorectal Dis 2007; 22: 183-9. [ Links ]
8. Halfdanarson TR, Rabe KG, Rubin J, Petersen GM. Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Oncol 2008; 19(10): 1727-33. [ Links ]
9. Hassan MM, Phan A, Li D, Dagohoy CG, Leary C, Yao JC. Risk factors associated with neuroendocrine tumors: A U.S.-based case-control study. Intern J Cancer 2008; 123(4): 867-73. [ Links ]
10. Vagefi PA, Razo O, Deshpande V, et al. Evolving patterns in the detection of pancreatic neuroendocrine tumors (PNETS): The Masschusetts General Hospital experience. Pancreas 2006; 33(4): 504 A. [ Links ]
11. Liu H, Zhang SZ, Wu YL, Fang HQ, Li JT, Sheng HW, et al. Diagnosis and surgical treatment of pancreatic endocrine tumors in 36 patients: a single-center report. Chin Med J 2007; 120(17): 1487-90. [ Links ]
12. Jagad RB, Koshariya M, Kawamoto J, Papastratis P, Kefalourous H, Patis V, et al. Pancreatic neuroendocrine tumors: our approach. Hepato-gastroenterology 2008; 55: 275-81. [ Links ]
13. Varas MJ. Síndrome de Zollinger-Ellison. Rev Med Univ Navarra 1995; 39: 18-23. [ Links ]
14. Varas MJ. Tumores endocrinos pancreáticos, ¿cuándo sospecharlos, cómo diagnosticarlos y cómo tratarlos? Revisiones en Gastroenterología 2000; 2(1): 47-55. [ Links ]
15. Metz DC, Jensen RT. Gastrointestinal neuroendocrine tumors: pancreatic endocrine tumors. Gastroenterology 2008; 135: 1469-92. [ Links ]
16. Kindmark H, Sundin A, Granberg D, Dunder K, Skogseid B, Janson ET, et al. Endocrine pancreatic tumors with glucagon hypersecretion: a retrospective study of 23 cases during 20 years. Med Oncol 2007; 24(3): 330-7. [ Links ]
17. Garbrecht N, Anlauf M, Schmitt A, Henopp T, Sipos B, Raffel A, et al. Somatostatin-producing neuroendocrine tumors of the duodenum and pancreas: incidence, types, biological behavior, association with inherited syndromes, and functional activity. Edocr Relat Cancer 2008; 15(1): 229-41. [ Links ]
18. Berna MJ, Hoffmann K, Long SH, Serrano J, Gibril F, Jensen RT. Serum gastrin in ZES: II. Prospective study of gastrin provocative testing in 293 patients from the National Institutes of Health and comparison with 537 cases from the literature. Evaluation of diagnostic critera, proposal of new criteria, and correlations with clinical and tumoral features. Medicine 2006; 85(6): 331-64. [ Links ]
19. Anderson MA, Carpenter S, Thompson NW, Nostrant TT, Elta GH, Scheiman JM. Endoscopic ultrasound is highly accurate ad directs managements in patients with neuroendocrine tumors of the pancreas. Am J Gastroenterol 2000; 95(9): 2271-7. [ Links ]
20. Varas MJ, Armengol JR, Boix J, et al. Diagnóstico y localización preoperatoria de los tumores endocrinos digestivos mediante ultrasonografía endoscópica. Gastroenterol y Hepatol 1999; 22: 223-6. [ Links ]
21. Varas MJ, Miquel JM, Maluenda MD, Boix J, Armengol JR. Preoperative detection of gastrointestinal neuroendocrine tumors using endoscopic ultrasonography. Rev Esp Enferm Dig 2006; 98: 828-36. [ Links ]
22. Varas MJ. Ultrasonografía endoscópica, aplicaciones diagnósticas y terapéuticas. Madrid: Ed. Médica Panamericana; 2008. [ Links ]
23. Santo E, Kariv R, Monges G, et al. The role of linear array endoscopic ultrasound with fine-needle aspiration in the diagnosis and preoperative evaluation of pancreatic neuroendocrine tumors -experience with 76 cases. Gastrointest Endosc 2002; 56 (4): S118. [ Links ]
24. Li Destri G, Regio E, Veroux M, Lanzafame S, Puleo S, Minutolo V. A rare cystic non-functioning neuroendocrine pancreatic tumor with an unusual presentation. Tumori 2006; 92: 260-3. [ Links ]
25. Ardengh SA, Komorowski RA, Demeure MJ, Wilson SD, Pitt HA. Cystic pancreatic neuroendocrine tumors: is preoperative diagnosis possible? J Gastrointest Surg 2002; 6: 66-74. [ Links ]
26. Baker MS, Knuth JL, DeWitt J, LeBlanc J, Cramer H, Howard TJ, Schmidt CM, et al. Pancreatic cystic neuroendocrine tumors: preoperative diagnosis with endoscopic ultrasound and fine-needle immunocytology. Gastrointest Surg 2008; 12(3): 450-6. [ Links ]
27. Kongkam P, Al-Haddad M, Attasaranya S, O'Neil J, Pais S, Sherman S, et al. EUS and clinical characteristics of cystic pancreatic neuroendocrine tumors. Endoscopy 2008; 40(7): 602-5. [ Links ]
28. Varas MJ, Tortosa F. Tumores endocrinos gastroenteropancreáticos y tratamiento con octreótida. Cirugía Española 1995; 58(4): 325-8. [ Links ]
29. Varas MJ, Gordillo J. Tumores pancreáticos endocrinos y terapia con interferón alfa 2b. Gastroenterol y Hepatol 1992; 15(7): 394-6. [ Links ]
30. Arnold R, Simon B, Wied M, Öberg K. Treatment of neuroendocrine GEP tumours with somatostatin analogues. Digestion 2000; 62 (Supl. 1): 84-97. [ Links ]
31. Pavel ME, Baum U, Hahn EG, Schuppan D, Lohmann T. Efficacy and tolerability of pegylated IFN-alpha in patients with neuroendocrine gastroenteropancreatic carcinomas. J Inteferferon Cytokine Res 2006; 26: 8-13. [ Links ]
32. Kulke MH, Stuart K, Enzinger PC, Ryan DP, Clark JW, Muzikansky A, et al. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol 2006; 24: 401-6. [ Links ]
33. Kulke MH, Lenz HJ, Meropol NJ, Posey J, Ryan DP, Picus J, et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol 2008; 26(20): 3403-10. [ Links ]
34. Norton JA, Fraker D, Alexander HR, Gibril F, Liewehr DJ, Venzon DJ, et al. Surgery increases survival in patients with gastrinoma. Ann Surg 2006; 244(3): 410-9. [ Links ]
35. Rothenstein J, Cleary SP, Pond GR, Dale D, Gallinger S, Moore MJ, et al. Neuroendocrine tumors of the gastrointestinal tract: A decade of experience at the Princess Margaret Hospital. Am J Clin Oncol 2008; 31(1): 64-70. [ Links ]
36. Ellison EC, Sparks J, Verducci JS, Johson JA, Muscarella P, Bllomston M, et al. 50-year appraisal of gastrinoma: recommendations for staging and treatment. J Am Coll Surg 2006; 202: 897-905. [ Links ]
37. Kazanjian KK, Reber HA, Hines OJ. Resection of pancreatic neuroendocrine tumors: results of 70 cases. Arch Surg 2006; 141: 765-9. [ Links ]
38. Böhmig M, Pape UF, Tiling N, et al. Prognostic factors in gastroenteropancreatic neuroendocrine tumors -a retrospective multivariate analysis. J Clin Oncol 2005; 23(16S): 4086. [ Links ]
39. Pape UF, Berndt U, Müller-Nordhorn J, Böhmig M, Roll S, Koch M, et al. Prognostic factors of long-term outcome in gastroenteropancreatic neuroendocrine tumours. Endocrine-Related Cancer 2008; 15(4): 1083-97. [ Links ]
40. Ekebland S, Skogseid B, Dunder K, Öberg K, Eriksson B. Prognostic factors and survival in 324 patients with pancreatic endocrine tumor treated at a single institution. Clin Cancer Research 2008; 14: 7798-03. [ Links ]
41. Bettini R, Boninsegna L, Mantovani W, Capelli P, Bassi C, Pederzoli P, et al. Prognostic factors at diagnosis and value of WHO classification in a mono-institucional series of 180 non-functioning pancreatic endocrine tumours. Ann Oncol 2008; 19(5): 903-8. [ Links ]
42. Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, et al. One hundred years after "carcinoid": epidemiology of and prognostic factors for neuroendocrine tumors in 35.825 cases in the United States. J Clin Oncol 2008; 26(18): 3063-72. [ Links ]
43. Hauso O, Gustafsson BI, Kidd M, Waldum HL, Drozdov I, Chan AK, et al. Neuroeendocrine tumor epidemiology: contrasting Norway and North America. Cancer 2008; 113(10): 2655-64. [ Links ]
44. Gustafsson BI, Kidd M, Chan A, Malfertheiner MV, Modlin IM. Bronchopulmonary neuroendocrine tumors. Cancer 2008; 113(1): 5-21. [ Links ]
45. Younes RN, on behalf of the GETNE. Neuroendocrine tumors: A registry of 1000 patients. Rev Assoc Med Bras 2008; 54(4): 305-7. [ Links ]
46. Martinez-Ares D, Souto J, Varas MJ, Espinós JC, Yáñez J, Abad R, et al. Endoscopic ultrasound-assisted endoscopic resection of carcinoid tumors of the gastrointestinal tract. Rev Esp Enferm Dig 2004; 96(12): 847-55. [ Links ]
47. Lips CJM, Lentjes EGWM, Höppener JWM. The spectrum of carcinoid tumours and carcinoid syndromes. Ann Clin Biochem 2003; 40: 612-27. [ Links ]
48. Ramaje JK, Davies AH, Ardill J, Bax N, Caplin M, Grossman A, et al.; UKNETwork for Neuroendocrine Tumours. Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours. Gut 2005; 54: iv1-iv16. [ Links ]
49. de Herder WW, O'Toole D, Rindi G, et al. ENETS consensus guidelines for the diagnosis and treatment of neuroendocrine gastrointestinal tumors: part 2 -midgut ad hindgut tumor. Neuroendocrinology 2008; 87: 1-63. [ Links ]
50. Halfdanarson TR, Rubin J, Farnell MB, Grant CS, Petersen GM. Pancreatic endocrine neoplasms: epidemiology and prognosis of pancreatic endocrine tumors. Endocrine-Related Cancer 2008; 15: 409-27. [ Links ]
51. Modlin IM, Oberg K, Chung DC, Jensen RT, de Herder WW, Thakker RV, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol 2008; 9: 61-72. [ Links ]
52. Jensen RT. Endocrine neoplasms of the pancreas. In: Yamada T, et al., editors. Textbook of Gastroenterology. 5th ed. Oxford: Blackwell; 2008. [ Links ]
53. Hill JS, McPhee JT, McDade TP, Zhou Z, Sullivan ME, Whalen GF, et al. Pancreatic neuroendocrine tumors: the impact of surgical resection on survival. Cancer 2009; 115(4): 741-51. [ Links ]
54. Strosberg J, Gardner N, Kvols L. Survival and prognostic factor análisis of 146 metastatic neuroendocrine tumors of the mid-gut. Neuroendocrinology 2009. [ Links ]
55. Lito P, Pantanowitz L, Marotti J, Aboulafia DM, Campbell V, Bower M, et al. Gastroenteropancreatic neuroendocrine tumors in patients with HIV infection: a trans-atlantic series (clinical investigation). Am J Med Sciences 2009; 337(1): 1-4. [ Links ]
56. Malagò R, D'Onofrio M, Zamboni GA, et al. Contrast-enhanced sonography of nonfunctioning pancreatic neuroendocrine tumors. AJR 2009; 192: 424-30. [ Links ]
57. Goudet P, Murat A, Cardet-Bauters C, et al. Thymic neuroendocrine tumors in multiple endocrine neoplasia type 1. Comparative study on 21 cases among a series of 761 MEN 1 from GTE (Group des Tumeurs Endocrines). World J Surg 2009; 26: 891-6. [ Links ]

