










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.107 no.1 Madrid ene. 2015
Acute abdomen in patients with systemic lupus erythematosus and antiphospholipid syndrome. Importance of early diagnosis and treatment
Abdomen agudo en paciente con lupus eritematoso sistémico y síndrome antifosfolípido. Importancia del diagnóstico y tratamiento precoz
Alberto Titos-García, José Manuel Aranda-Narváez, Naira Marín-Camero, Isabel Rosa Fernández-Burgos, María Custodia Montiel-Casado, Antonio Jesús González-Sánchez and Julio Santoyo-Santoyo
Department of General, Digestive and Transplantation Surgery. Hospital Regional Universitario de Málaga. Málaga, Spain
ABSTRACT
Systemic lupus erithematosus (SLE) is an autoimmune disease with multiorgan involvement caused principally by vasculitis of small vessels. The gastrointestinal tract is one of the most frequently affected by SLE, with abdominal pain as the most common symptom. An early diagnosis and treatment of lupus enteritis is essential to avoid complications like hemorrhage or perforation, with up to 50 % of mortality rate. However, differential diagnosis sometimes is difficult, especially with other types of gastrointestinal diseases as digestive involvement of antiphospholipid syndrome (APS), moreover when both entities may coexist. We describe the case of a patient with both diseases that was diagnosed with lupus enteritis and treated with steroid therapy; the patient had an excellent response.
Key words: Systemic lupus erithematosus. Antiphospholipid syndrome. Lupus enteritis. Vasculitis. Steroids.
RESUMEN
El lupus eritematoso sistémico es una enfermedad autoinmune con afectación multivisceral causada principalmente por vasculitis de pequeño vaso. El tracto gastrointestinal es uno de los órganos más frecuentemente afectados, siendo el dolor abdominal el síntoma predominante. La enteritis lúpica requiere un diagnóstico y tratamiento precoces para evitar complicaciones como la hemorragia digestiva y la perforación intestinal, que pueden alcanzar una mortalidad de hasta el 50 %. Su diagnóstico a veces se ve dificultado por la presencia de otras patologías con afectación gastrointestinal similar como ocurre en el síndrome antifosfolípido. Presentamos el caso de una paciente con ambas enfermedades que fue diagnosticada de enteritis lúpica y tratada de forma conservadora con terapia corticoidea de choque. La paciente tuvo una respuesta excelente al tratamiento.
Palabras clave: Lupus eritematoso sistémico. Síndrome antifosfolípido. Enteritis lúpica. Vasculitis. Esteroides.
Introduction
Systemic lupus erithematosus (SLE) and antiphospholipid syndrome (APS) are two autoimmune diseases with multiorgan involvement caused principally by vasculitis of small vessels. However, pathophysiology is different in both diseases. The vasculopathy that characterizes SLE is an inflammatory type secondary to immuno-complex deposits in vessel walls, meanwhile APS is characterized by a state of hypercoagulability due to the presence of autoantibodies, potentially resulting in thrombosis. Historically, APS was described as a clinical manifestation of SLE. Since 1985, several studies have reported differences between both diseases, and so APS must be considered as a different entity (1,2).
Gastrointestinal manifestations are relatively frequent in patients with SLE, reaching up to 40 % in cases of active disease. Symptoms may vary from unspecific abdominal pain to life-threatening acute abdomen, known as mesenteric vasculitis or lupus enteritis (LE). Early diagnosis and treatment are crucial to avoid complications as hemorrhage, necrosis or intestinal perforation with a high mortality of up to 50 % (2,3).
On the other side, gastrointestinal involvement is rare in APS (approximately 1.5 % of patients), and includes predominantly affectation of the liver and spleen. However, it is necessary to bear APS in mind in differential diagnosis of acute abdomen, since it may cause a massive intestinal thrombosis known as Asherson's syndrome (4).
We describe the case of a patient with SLE and APS, with abdominal pain and semiology compatible with acute abdomen that was diagnosed with lupus enteritis and treated medically with steroid therapy, resulting in a rapid improvement of her gastrointestinal symptoms.
Case report
A 55-year-old woman, with previous diagnosis (40 years before) of SLE with involvement of the kidney, central nervous system and skin, had a concomitant APS with peripheral vasculopathy and amputation of both inferior limbs. She was admitted with abdominal pain, nausea, vomiting, diarrhea, abdominal distension and anorexia during the last two weeks, with worsening during the three days previous to her admission. Neither fever nor other symptoms suggesting activity of SLE were referred. Physical exploration revealed distended abdomen with tympanism, absence of bowel sounds and diffuse abdominal pain with unclear signs of peritoneal affectation (defense or rebounding sign). Laboratory tests showed leucopenia (3 x 103/mm3) and thrombocytopenia (99 x 103/mm3). Abdominal computed tomography (CT) findings were dilated small bowel, marked edematous thickening of the wall of the right colon and small bowel, and a mild amount of ascites. A diagnosis of intestinal ischemia probably related with APS was established and medical treatment was indicated, consisting basically on intestinal rest, intravenous fluids, non-steroid anti-inflammatory drugs (NSAIDs) and therapeutic doses of low-weight subcutaneous heparin. Initially, surgical exploration was discarded due to the extended period of duration of symptoms and the lack of clear signs of acute abdomen.
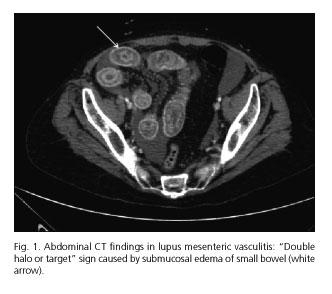
Further laboratory data revealed persistence of leucopenia and thrombocytopenia, decreased complement factors (C3 and C4) and positive antinuclear antibodies (ANA), anticardiolipin, antiB2 GP1 and lupus anticoagulant. After four days without improvement of symptoms and persistence of dilatation of the lumen of the small bowel on abdominal X-ray, indication of a second CT was established, which showed similar but more pronounced findings if compared with the previous one: Dilated bowel, marked thickening of the wall of jejunum and ileum caused by edema ("double halo or target sign"), mesenteric edema, engorgement of mesenteric vessels and moderate ascites. Abdominal exploration had worsened too, showing clinical signs of acute abdomen, and surgeon on duty was called. Considering clinical profile, radiological findings and laboratory data, the possibility of LE was then suggested and extensively discussed, and intravenous administration of high doses of corticosteroids was initiated. She was treated with 1 g/d of 6-methylprednisolone (6-MP) during 48 hours and then reduced to 500 mg/d. Improvement of her symptoms was visible during the first 24 hours, and after three days the patient was asymptomatic and allowed us to reintroduce oral diet. Previous malnutrition and a respiratory infection requiring specific antibiotic therapy prolonged hospital stay, but patient was discharged 29 days later with a maintenance dose of 60 mg/d of prednisone with progressive withdrawal.
Discussion
Systemic lupus erithematosus (SLE) is a chronic inflammatory disease that belongs to the group of the diseases known as systemic vasculitis whose common pathologic feature is inflammation of the walls of small vessels, with other diseases like polyarteritis nodosa (PAN), Churg-Strauss syndrome, Wegener granulomatosis (WG) or Takayasu arteritis. SLE is characterized by the involvement of various organs, frequently skin, kidney, central nervous system and gastrointestinal tract. Unspecific gastrointestinal symptoms are frequently observed in this disease; anorexia, nausea or vomiting may be present in approximately 30-50 % of patients, being abdominal pain the most common symptom (around 20 %) (5-7).
Lupus mesenteric vasculitis (LMV) is one of the most serious complications of SLE, with an estimated incidence ranging from 0.2 to 9.7 % of patients with active disease and less than 1 % of those ones with inactive disease. It is unusual that LMV constitutes the initial presentation of SLE. It is frequently associated with thrombocytopenia, lymphopenia and central nervous system and skin involvement. Triggering factors of LMV are unknown, but bacterial infections (associated with changes of the intestinal flora), animal viruses, cytomegalovirus infection, eosinophilia, chemicals, NSAIDs, metallic particles, caffeine, exercise and certain foods and herbal medicines have been proposed as potential etiologic factors (2,8). LMV affects preferentially the territory of superior mesenteric artery, especially jejunum and ileum (80-85 %), and it is produced by accumulation of immunocomplex deposits in the wall of the vessels (5). Although generally shows a benign and insidious clinical course, sometimes LMV may produce severe intestinal ischemia complicated with hemorrhage, ulceration and perforation, with a mortality that may reach up to 50 % (2,9).
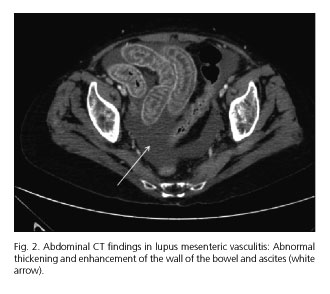
Early diagnosis and treatment are crucial to avoid these catastrophic complications. Contrast-enhanced CT is considered the most sensitive and specific noninvasive tool for diagnosis of LMV. Most common CT findings are segmental or multifocal bowel involvement alternating with normal segments of healthy bowel, bowel dilatation, engorgement of mesenteric vessels, mesenteric edema, bowel wall thickening with abnormal enhancement caused by submucosal edema called "double halo or target sign" (Fig. 1) and ascites (Fig. 2). Other CT findings suggestive of LMV are hepatosplenomegaly, retroperitoneal lymphadenopathy and pancreatitis (5-10). Ultrasonography is another noninvasive tool which may be helpful in both diagnosis and follow-up of LMV. Magnetic resonance imaging (MRI), double-contrast radiographic assessment of the upper and lower gastrointestinal tract, capsule endoscopy and Ga67 gammagraphy have shown to be useful, but these techniques have not been accepted widely. As affected vessels are usually deep and inaccessible, endoscopy-guided biopsy is not recommended for definitive diagnosis (2,5).
The mechanism of injury is an inflammatory but not an atherosclerotic or embolic occlusion of small vessels. This is the reason why medical treatment is based on aggressive doses of intravenous corticosteroids and absolute intestinal rest, followed by a gradual steroids withdrawal and reintroduction of oral nutrition. Another option is prescription of cyclophosphamide (initial doses of 1-2 mg/kg/d and afterwards a monthly dose) or mycophenolatemofetil, but several authors recommend this therapy only in case of failure of steroid therapy. Finally, surgical evaluation and early laparotomy must be indicated if medical treatment is not successful (within 48 hours), because prognosis of these patients might be improved. Ultrasonography and abdominal CT are useful tools in follow-up of medical therapy. A combination of cyclophosphamide and corticosteroids must be prescribed if there is evidence of active disease after surgery (2,5-8). In recurrent forms micofenolate mofetil, azathioprine, ciclophosphamide and rituximab have been successfully used to prevent further recurrence in a limited numbers of patients.
APS is an autoimmune disease characterized by recurrent arterial and venous thrombosis. Patients with APS show high levels of antiphospholipid antibodies (anti-cardiolipin, antiB2 GP1 antibodies and positive lupus anticoagulant). APS may be defined as primary in about 53 % of cases, but it may be associated with other autoimmune disease such as lupus, and then it must be defined as secondary. In fact, initially APS was described in a group of patients with SLE (secondary disease), but since 1985 primary APS must be considered an independent disease (1,4,11,12). It is a multiorgan disease, with similar clinical profile when compared with lupus: affectation of central nervous system (migraine, epilepsy, cognitive disorders), pulmonary (alveolar hemorrhage), hematological (thrombocytopenia, autoimmune hemolytic anemia), renal (glomerulonephritis), skin (livedo reticularis and cutaneous ulcers), recurrent fetal loss (1,13). Nevertheless, in APS is infrequent that gastrointestinal tract is affected (about 1.5 %), and when APS involves digestive system this affectation includes principally hepatic manifestations (Budd-Chiari syndrome, hepatic venous thrombosis), splenic or pancreatic infarction. Intestinal infarction is rare, but a catastrophic variant known as Asherson syndrome has been previously described. It is an accelerated form of APS resulting in diffuse intestinal infarction and multiorgan failure because of multiple small vessel occlusions, with up to 50 % of mortality rate. The most common precipitating conditions are infections (22 %) and surgical procedures (10 %). Other less common causes are anticoagulation withdrawal or low international normalized ratio (INR), medications, obstetric complications and neoplasia. Radiological findings (abdominal CT) in these patients are similar to other entities that lead to intestinal ischemia: enhancement of the wall of the bowel, free intraabdominal fluid, lack of enhancement of the arterial vasculature with intravenous contrast, intestinal pneumatosis and gas in the portal system. Medical support is a combination of intravenous glucocorticoids and anticoagulant therapy, associating initially low-molecular-weight heparin and warfarin and ciclophosphamide in cases with concomitant SLE. Duration of treatment depends on clinical course and an optimal length has not been well defined. Surgery must be reserved for cases of failure of medical treatment or presence of obvious peritonitis. Hydroxychloroquine, plasmapheresis and other immunomodulatory therapies (through a decrease of levels of anticardiolipin antibodies) have been purposed by several authors for thrombosis prevention in these patients (1,4,5,14,15).
In our patient, the initial non-specific symptoms led us to a wrong diagnosis and therefore the beginning of steroid therapy was delayed. In spite of that, it is important to emphasize that the patient had a previous diagnosis of both entities, SLE and APS, and both were potentially responsible of the clinical profile. As soon as radiological CT images suggested the possibility of LMV we focused on it and an appropriate therapy was prescribed, but this delay could cause complications as hemorrhage or perforation with an associated high morbimortality. Immediate response to medical therapy with steroids confirmed the diagnosis.
In summary, SLE and APS are two different autoimmune diseases with a few similar characteristics, and they may be present in the same patient concomitantly. Among 4-10 % of patients with APS will develop a fully established SLE in the future. Gastrointestinal affectation is more frequent in SLE, although both diseases may be the origin of an acute abdomen with a high rate of mortality. Therefore, a diagnostic effort must be performed in order to establish an adequate medical treatment, trying to avoid an undesirable medical course and surgery. Contrast-enhaced abdominal CT is considered the most useful tool for diagnosis of mesenteric vasculitis. Classical image "in reveille or double halo" suggests strongly the diagnosis of SLE, meanwhile CT findings of gastrointestinal affectation of APS are usually unspecific. Medical treatment must be considered the first option in both cases, with high doses of intravenous glucocorticoids in SLE, and glucocorticoids with anticoagulants in APS. Surgery must be reserved for cases without successful medical treatment or complications.
References
1. Shoenfeld Y, Meroni PL, Toubi E. Antiphospholipid syndrome and systemic lupus erythematosus: Are they separate entities or just clinical presentations on the same scale?. Curr Opin Rheumatol 2009;21:495-500. [ Links ]
2. Ju JH, Min JK, Jung CK, Oh SN, Kwok SK, Kang KY, et al. Lupus mesenteric vasculitis can cause acute abdominal pain in patients with SLE. Nat Rev Rheumatol 2009;5:273-81. [ Links ]
3. Makary R, Davis C, Shuja S. Medium and small vessel vasculitis with large bowel infarction in systemic lupus erythematosus: A case report. Am J Gastroenterol 2009;104:1859-60. [ Links ]
4. Bachmeyer C, Barrier A, Frazier A, Fulgencio JP, Lecomte I, Grateau G, et al. Diffuse large and small bowel necrosis in catastrophic antiphospholipid syndrome. Eur J Gastroenterol Hepatol 2006;18:1011-4. [ Links ]
5. Passam FH, Diamantis ID, Perisinaki G, Saridaki Z, Kritikos H, Georgopoulos D, et al. Intestinal ischemia as the first manifestation of vasculitis. Semin Arthritis Rheum 2004;34:431-41. [ Links ]
6. Kaneko Y, Hirakata M, Suwa A, Satoh S, Nojima T, Ikeda Y, et al. Systemic lupus erythematosus associated with recurrent lupus enteritis and peritonitis. Clin Rheumatol 2004;23:351-4. [ Links ]
7. Chung H, Ramji A, Davis J, Chang S, Reid G, Salh B, et al. Abdominal pain as the initial and sole clinical presenting feature of systemic lupus erythematosus. Can J Gastroenterol 2003;17:111-3. [ Links ]
8. Janssens P, Arnaud L, Galicier L, Mathian A, Hie M, Sene D, et al. Lupus enteritis: From clinical findings to therapeutic management. Orphanet J Rare Dis 2013;8:67. [ Links ]
9. Assimakopoulos SF, Dimitropoulou D, Liossis SC. Gastrointestinal: Intestinal vasculitis associated with systemic lupus erythematosus. J Gastroenterol Hepatol 2008;23:992. [ Links ]
10. Huang DF, Chen WS. Lupus-associated intestinal vasculitis. N Engl J Med 2009;361:3. [ Links ]
11. Patel YI, John A, McHugh NJ. Antiphospholipid syndrome with proliferative vasculopathy and bowel infarction. Rheumatology (Oxford) 2000;39:108-10. [ Links ]
12. Kato N, Yukioka H, Yoshida G, Kurita S, Hirakawa K, Koh KR. Superior mesenteric vein thrombosis presented transient false positivity for lupus anticoagulant under heparin treatment. Eur J Emerg Med 2002;9:67-70. [ Links ]
13. Uthman I, Khamashta M. The abdominal manifestations of the antiphospholipid syndrome. Rheumatology 2007;46:1641-7. [ Links ]
14. Cervera R, Bucciarelli S, Plasín MA, Gómez-Puerta JA, Plaza J, Pons-Estel G, et al. Catastrophic antiphospholipid síndrome (CAPS): Descriptive analysis of a series of 280 patients from the "CAPS Registry". J Autoimmun 2009;32:240-5. [ Links ]
15. Cervera R. CAPS Registry. Lupus 2012;21:755-7. [ Links ]
![]() Correspondence:
Correspondence:
Alberto Titos García.
Secretaría del Servicio de Cirugía General y Digestiva.
Hospital Regional Universitario de Málaga.
Avenida de Carlos Haya, s/n.
29010 Málaga, Spain
e-mail:
albertotitosg@hotmail.com
Received: 28-03-2014
Accepted: 07-04-2014

