










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.107 no.1 Madrid ene. 2015
Abdomen agudo en paciente con lupus eritematoso sistémico y síndrome antifosfolípido. Importancia del diagnóstico y tratamiento precoz
Acute abdomen in patients with systemic lupus erythematosus and antiphospholipid syndrome. Importance of early diagnosis and treatment
Alberto Titos García, José Manuel Aranda Narváez, Naira Marín Camero, Isabel Rosa Fernández Burgos, María Custodia Montiel Casado, Antonio Jesús González Sánchez y Julio Santoyo Santoyo
Servicio de Cirugía General, Digestiva y Trasplantes. Hospital Regional Universitario de Málaga. Málaga
Dirección para correspondencia
RESUMEN
El lupus eritematoso sistémico es una enfermedad autoinmune con afectación multivisceral causada principalmente por vasculitis de pequeño vaso. El tracto gastrointestinal es uno de los órganos más frecuentemente afectados, siendo el dolor abdominal el síntoma predominante. La enteritis lúpica requiere un diagnóstico y tratamiento precoces para evitar complicaciones como la hemorragia digestiva y la perforación intestinal, que pueden alcanzar una mortalidad de hasta el 50 %. Su diagnóstico a veces se ve dificultado por la presencia de otras patologías con afectación gastrointestinal similar como ocurre en el síndrome antifosfolípido. Presentamos el caso de una paciente con ambas enfermedades que fue diagnosticada de enteritis lúpica y tratada de forma conservadora con terapia corticoidea de choque. La paciente tuvo una respuesta excelente al tratamiento.
Palabras clave: Lupus eritematoso sistémico. Síndrome antifosfolípido. Enteritis lúpica. Vasculitis. Esteroides.
ABSTRACT
Systemic lupus erithematosus (SLE) is an autoimmune disease with multiorgan involvement caused principally by vasculitis of small vessels. The gastrointestinal tract is one of the most frequently affected by SLE, with abdominal pain as the most common symptom. An early diagnosis and treatment of lupus enteritis is essential to avoid complications like hemorrhage or perforation, with up to 50 % of mortality rate. However, differential diagnosis sometimes is difficult, especially with other types of gastrointestinal diseases as digestive involvement of antiphospholipid syndrome (APS), moreover when both entities may coexist. We describe the case of a patient with both diseases that was diagnosed with lupus enteritis and treated with steroid therapy; the patient had an excellent response.
Key words: Systemic lupus erithematosus. Antiphospholipid syndrome. Lupus enteritis. Vasculitis. Steroids.
Introducción
El lupus eritematoso sistémico (LES) y el síndrome antifosfolípido (SAF) son dos enfermedades autoinmunes con afectación multiorgánica causada por vasculopatía de pequeño vaso. El mecanismo fisiopatológico es distinto en ambas enfermedades. En el LES la afectación se produce por fenómenos inflamatorios secundarios al depósito de inmunocomplejos en las paredes de los vasos; sin embargo, en el SAF se debe a fenómenos trombóticos producidos por un estado de hipercoagulabilidad relacionado con la presencia de autoanticuerpos. Históricamente, el SAF era descrito como una manifestación clínica del LES. No fue hasta 1985 cuando varios estudios reportaron diferencias entre ambas enfermedades, siendo considerado desde entonces el SAF como una entidad diferente (1,2).
La afectación del tracto gastrointestinal es relativamente frecuente en los pacientes con LES, llegando hasta el 40 % en los casos de enfermedad activa. La clínica varía desde dolor abdominal inespecífico hasta un cuadro de abdomen agudo llamado vasculitis mesentérica o enteritis lúpica (EL). En este último caso el diagnóstico y el tratamiento precoz es fundamental para evitar complicaciones como hemorragia, necrosis o perforación intestinal con una mortalidad cercana al 50 % de los pacientes (2,3).
Por el contrario, la aparición de clínica digestiva en el SAF es rara, alrededor del 1,5 % de los pacientes, y afecta principalmente a hígado y bazo. A pesar de esto hay que tener en cuenta el SAF en el diagnóstico diferencial del abdomen agudo, ya que en ocasiones puede producir una trombosis masiva intestinal conocido como síndrome de Asherson, difícil de diferenciar de la EL y con un manejo distinto (4).
Presentamos el caso de una paciente con historia de LES de larga evolución y SAF, ingresada en nuestro hospital con diagnóstico de EL que fue tratada mediante terapia esteroidea, resultando en una rápida mejoría de sus síntomas gastrointestinales.
Caso clínico
Mujer de 55 años, con diagnóstico de LES desde hace 40 años (afectación renal, nerviosa y cutánea), SAF y amputación supracondílea de ambos miembros inferiores por vasculopatía periférica secundaria. Consultó en urgencias de nuestro hospital por un cuadro de dolor abdominal difuso de dos semanas de evolución, náuseas, vómitos y anorexia, que habían aumentado en los últimos días. Refería disminución del tránsito intestinal, con escasas deposiciones de carácter diarreico. No presentaba fiebre ni otros síntomas de actividad del LES. La exploración física reveló un abdomen distendido y timpánico, con ausencia de ruidos intestinales y dolor abdominal difuso sin signos claros de irritación peritoneal. En la analítica de ingreso destacaba leucopenia (3,0x109/L) y trombopenia (99x109/L). Se realizó una tomografía computerizada (TC) de abdomen en la que se observaba engrosamiento de la pared del colon ascendente e íleon terminal, con moderada cantidad de líquido libre. Con el diagnóstico de sospecha de isquemia intestinal segmentaria probablemente relacionada con su SAF fue ingresada en el hospital, instaurándose tratamiento conservador con reposo intestinal, fluidos intravenosos, antiinflamatorios no esteroideos y dosis terapéutica de heparina de bajo peso molecular. Inicialmente, una exploración quirúrgica fue descartada debido a la duración extensa de los síntomas y la falta de signos claros de abdomen agudo.
En analíticas posteriores se evidenció persistencia de la leucopenia y la trombopenia, disminución de los factores del complemento (C3 de 66 mg/dL, C4 de 6 mg/dL) y anticuerpos ANA, anticardiolipina, antiB2 GP1 y anticoagulante lúpico positivos. Tras cuatro días de ingreso sin mejoría de los síntomas y persistencia de dilatación de intestino delgado en las radiografías de abdomen se decidió realizar nueva TC de abdomen, que mostró engrosamiento y edematización de asas intestinales a nivel de yeyuno e íleon pélvico con imagen de "doble halo o diana", dilatación proximal de las asas de delgado, ingurgitación del meso y mayor cantidad de líquido libre. La exploración física había empeorado, mostrando signos de abdomen agudo evidentes. Considerando el perfil clínico, los hallazgos radiológicos y datos de laboratorio, la posibilidad de una enteritis lúpica fue sugerida y extensamente debatida, iniciándose la administración intravenosa de altas dosis de corticosteroides. Fue tratada con 1 g/d de 6-metillprednisolona (6-MP) durante las primeras 48 horas y después con 500 mg/d. La mejoría clínica fue evidente en las primeras 24 horas, y después de tres días la paciente estaba asintomática, lo que permitió reintroducir la dieta oral. El estado de malnutrición previo y una infección respiratoria requirieron antibioterapia específica que prolongó la estancia hospitalaria. La paciente fue dada de alta 29 días después con una dosis de mantenimiento de 60 mg/d de prednisona con retirada progresiva.
Discusión
El lupus eritematoso sistémico es una enfermedad crónica inflamatoria perteneciente al grupo de las enfermedades conocidas como vasculitis sistémicas, cuya característica común es la inflamación de las paredes de los vasos sanguíneos, en este caso de pequeño calibre. Dentro de este grupo están la poliarteritis nodosa (PAN), el síndrome de Churg-Strauss, la granulomatosis de Wegener (GW) o la arteritis de Takayasu. El LES se caracteriza por una afectación multivisceral, fundamentalmente piel, riñones, sistema nervioso central y otros órganos como el tracto gastrointestinal. La aparición de clínica abdominal inespecífica es bastante frecuente en estos pacientes; anorexia, náuseas o vómitos pueden darse hasta en un 30-50 % de los casos, siendo el dolor abdominal el más frecuente, aproximadamente en un 20 % (5-7).
La vasculitis mesentérica o enteritis lúpica es una de las complicaciones más graves del LES, con una prevalencia del 0,2 al 9,7 % en pacientes con enfermedad activa y menor del 1 % con enfermedad inactiva. Es inusual que la EL constituya el síntoma de debut de un LES. Está frecuentemente asociado a trombocitopenia, afectación nerviosa y cutánea, y linfopenia. Los factores desencadenantes son desconocidos, aunque las infecciones bacterianas con alteración de la flora intestinal, infección por citomegalovirus, eosinofilia, determinados medicamentos, partículas metálicas, virus animales, cafeína, ejercicio y determinadas hierbas medicinales han sido definidas como factores predisponentes (2,8). Suele afectar preferentemente al territorio de la arteria mesentérica superior, sobre todo yeyuno e íleon en el 80-85 % (5) y está producida por el depósito de inmunocomplejos en las paredes de los vasos sanguíneos. Aunque generalmente presenta un curso clínico benigno e insidioso, en ocasiones la EL puede provocar una isquemia intestinal severa complicada con hemorragia, ulceraciones e incluso perforaciones, con una mortalidad de hasta el 50 % (2,9).
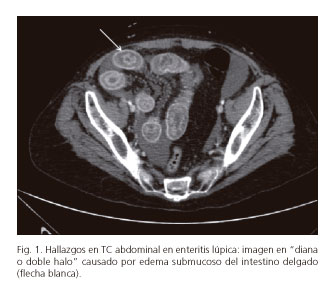
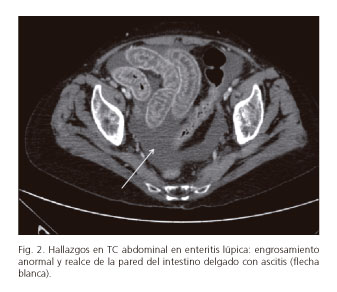
El diagnóstico precoz y el tratamiento es crucial para evitar estas complicaciones. El TC de abdomen con contraste está considerada la herramienta no invasiva más sensible y específica para el diagnóstico de EL. Los hallazgos más comunes en estos pacientes son afectación segmentaria intestinal alternando con zonas de intestino sano, dilatación de asas, ingurgitación de los vasos mesentéricos con engrosamiento del meso adyacente, engrosamiento difuso de la pared intestinal causado por el edema submucoso del asa, produciendo el signo de "doble halo o diana" (Fig. 1), y ascitis (Fig. 2). Otros datos inespecíficos del TC que sugieren EL son hepatoesplenomegalia, linfadenopatías retroperitoneales y pancreatitis aguda (5-10). La ultrasonografía es otra prueba no invasiva que, aunque menos sensible, ha demostrado ser útil tanto en el diagnóstico como en el seguimiento de esta patología. La resonancia magnética (RMN), el tránsito intestinal, la cápsula endoscópica y la gammagrafia con Ga67 han demostrado ser útiles, pero no son pruebas que puedan realizarse de rutina y por tanto no están todavía ampliamente utilizadas. La biopsia guiada por endoscopia o colonoscopia no suele ser concluyente ya que la afectación de los vasos suele ser profunda e inaccesible (2,5).
El mecanismo de la lesión es inflamatorio, no una oclusión ateroesclerótica o embólica. Esta es la razón de que el tratamiento esté basado en dosis agresivas de corticosteroides IV (dosis inicial de 1-2 mg/kg/d) y reposo intestinal absoluto, seguido de un descenso gradual de la dosis de esteroides y una reintroducción cuidadosa de la tolerancia digestiva. Otra opción que ha demostrado ser útil es el tratamiento con ciclofosfamida (dosis inicial 1-2 mg/kg/d y posteriormente una dosis mensual) o con micofenolato mofetil; sin embargo, algunos autores prefieren dejar esta terapia como segunda línea en caso de fracaso del tratamiento con esteroides. Finalmente, la evaluación quirúrgica y una laparotomía precoz (dentro de las primeras 48 horas) deben ser indicadas en caso de fracaso del tratamiento médico, dado que mejora el pronóstico del enfermo. En el seguimiento de los pacientes tanto la TC como la US son de gran utilidad, observando la desaparición del edema submucoso intestinal normalmente a la semana de iniciado el tratamiento médico. El pronóstico del paciente va a depender de la extensión de intestino afectado y del momento en que se realice la intervención quirúrgica. El tratamiento combinado con ambos inmunosupresores es eficaz para control de la enfermedad tras cirugía intestinal (2,5-8). En formas recurrentes el micofenolato mofetil, la azatioprina, la ciclofosfamida y el rituximab han tenido éxito en la prevención de la recurrencia en un número limitado de pacientes.
El SAF es una enfermedad autoinmune caracterizada por episodios de trombosis arterial y venosa. Los pacientes con SAF presentan niveles elevados de anticuerpos antifosfolípido (anticardiolipina, anticoagulante lúpico y anti-B2 GP1). Puede desarrollarse bien como enfermedad única, denominándose SAF primario (53 % de los casos), o bien puede aparecer asociada a otras enfermedades autoinmunes como el lupus, denominándose entonces SAF secundario (40 % de los casos). De hecho, los primeros casos que se dieron a conocer de esta enfermedad fueron descubiertos en pacientes con LES, de ahí que inicialmente se pensara que formaba parte de esta enfermedad. No fue hasta 1985 cuando se definió el SAF primario como entidad independiente (1,4,11,12). Es una enfermedad multiorgánica, con un perfil clínico similar cuando lo comparas con el LES: afectación del sistema nervioso central (migraña, epilepsia, desórdenes cognitivos), pulmonar (hemorragia alveolar), hematológico (trombocitopenia, anemia hemolítica autoinmune), renal (glomerulonefritis), cutáneo (lívedo reticularis y úlceras cutáneas), abortos fetales de repetición, necrosis avascular ósea (1,13). Sin embargo, al contrario de lo que ocurre en el LES, la afectación del tracto gastrointestinal es rara, en torno al 1,5 %, e incluye principalmente manifestaciones hepáticas (síndrome de Budd Chiari, trombosis venosa hepática), infarto esplénico y pancreático. El infarto intestinal es raro y da lugar a un cuadro catastrófico conocido como síndrome de Asherson. Es una forma acelerada de SAF que resulta en un infarto intestinal masivo y fallo multiorgánico por oclusión trombótica de pequeños vasos intestinales, alcanzando hasta un 50 % de mortalidad. Las condiciones precipitantes más comunes son infecciones (22 %) y procedimientos quirúrgicos (10 %). Otras causas menos comunes son cese o ineficacia de anticoagulación, medicación, complicaciones obstétricas y neoplasias. Los hallazgos radiológicos (TC) en estos pacientes son similares a otras entidades que producen isquemia intestinal en mayor o menor grado de severidad: engrosamiento de las asas del territorio del vaso afecto, líquido libre intraabdominal, cese o disminución del flujo a nivel de lo vasos mesentéricos afectados y en cuadros avanzados pneumatosis intestinal y gas en el sistema portal. El tratamiento consiste en la administración simultánea de glucocorticoides IV y terapia anticoagulante, asociando inicialmente heparina de bajo peso molecular y warfarina hasta obtener un INR de 2,5-3,5; y ciclofosfamida en los casos de LES concomitante. La duración del tratamiento depende de la evolución clínica del paciente, aunque no está bien definida. La cirugía se reserva en el caso de fracaso del tratamiento médico o peritonitis abdominal franca. La hidroxicloroquina, plasmaféresis y otras terapias inmunomoduladoras, que actúan reduciendo los niveles de anticuerpos anticardiolipina, han sido propuestas por algunos autores para la prevención trombótica en estos pacientes (1,4,5,14,15).
En el caso de nuestra paciente, la clínica inespecífica inicial nos llevó a un error diagnóstico y por tanto a una demora en el inicio de la terapia esteroidea. Además de esto, es importante enfatizar que la paciente tenía el diagnóstico previo de las dos enfermedades, LES y SAF, y que ambas podían ser potencialmente responsables del cuadro clínico. Tan pronto como las imágenes del TC sugirieron la posibilidad de EL se inició la terapia adecuada, aunque el retraso en el diagnóstico pudo haber causado complicaciones como hemorragia o perforación con una alta morbimortalidad asociada. Finalmente, la respuesta inmediata al tratamiento médico con corticoides confirmó el diagnóstico.
Discusión
El LES y el SAF son dos enfermedades autoinmunes independientes con algunas características similares, y pueden aparecer juntas en el mismo paciente. Alrededor del 4-10 % de los pacientes con SAF desarrollarán un LES plenamente establecido en el futuro. La afectación gastrointestinal es más frecuente en el LES, aunque ambas enfermedades pueden producir cuadros abdominales agudos con una alta tasa de mortalidad. Es fundamental en estos pacientes establecer el diagnóstico precoz y el tratamiento correcto, ya que de ello dependerá la evolución y la necesidad de cirugía. El TC de abdomen con contraste está considerada la herramienta más útil para el diagnóstico de las vasculitis mesentéricas. La imagen clásica "en diana o doble halo" sugiere fuertemente el diagnóstico de LES, mientras que los hallazgos de afectación gastrointestinal del SAF suelen ser inespecíficos. El tratamiento médico debe ser considerado como primera opción en ambos casos, con altas dosis de glucocorticoides intravenosos en el LES, y glucocorticoides con anticoagulantes en el SAF. La cirugía debe ser reservada para casos sin éxito en el tratamiento médico o aparición de complicaciones.
![]() Dirección para correspondencia:
Dirección para correspondencia:
Alberto Titos García.
Secretaría del Servicio de Cirugía General y Digestiva.
Hospital Regional Universitario de Málaga.
Avenida de Carlos Haya, s/n.
29010 Málaga
e-mail:
albertotitosg@hotmail.com
Recibido: 28-03-2014
Aceptado: 07-04-2014
Bibliografía
1. Shoenfeld Y, Meroni PL, Toubi E. Antiphospholipid syndrome and systemic lupus erythematosus: Are they separate entities or just clinical presentations on the same scale?. Curr Opin Rheumatol 2009;21:495-500. [ Links ]
2. Ju JH, Min JK, Jung CK, Oh SN, Kwok SK, Kang KY, et al. Lupus mesenteric vasculitis can cause acute abdominal pain in patients with SLE. Nat Rev Rheumatol 2009;5:273-81. [ Links ]
3. Makary R, Davis C, Shuja S. Medium and small vessel vasculitis with large bowel infarction in systemic lupus erythematosus: A case report. Am J Gastroenterol 2009;104:1859-60. [ Links ]
4. Bachmeyer C, Barrier A, Frazier A, Fulgencio JP, Lecomte I, Grateau G, et al. Diffuse large and small bowel necrosis in catastrophic antiphospholipid syndrome. Eur J Gastroenterol Hepatol 2006;18:1011-4. [ Links ]
5. Passam FH, Diamantis ID, Perisinaki G, Saridaki Z, Kritikos H, Georgopoulos D, et al. Intestinal ischemia as the first manifestation of vasculitis. Semin Arthritis Rheum 2004;34:431-41. [ Links ]
6. Kaneko Y, Hirakata M, Suwa A, Satoh S, Nojima T, Ikeda Y, et al. Systemic lupus erythematosus associated with recurrent lupus enteritis and peritonitis. Clin Rheumatol 2004;23:351-4. [ Links ]
7. Chung H, Ramji A, Davis J, Chang S, Reid G, Salh B, et al. Abdominal pain as the initial and sole clinical presenting feature of systemic lupus erythematosus. Can J Gastroenterol 2003;17:111-3. [ Links ]
8. Janssens P, Arnaud L, Galicier L, Mathian A, Hie M, Sene D, et al. Lupus enteritis: From clinical findings to therapeutic management. Orphanet J Rare Dis 2013;8:67. [ Links ]
9. Assimakopoulos SF, Dimitropoulou D, Liossis SC. Gastrointestinal: Intestinal vasculitis associated with systemic lupus erythematosus. J Gastroenterol Hepatol 2008;23:992. [ Links ]
10. Huang DF, Chen WS. Lupus-associated intestinal vasculitis. N Engl J Med 2009;361:3. [ Links ]
11. Patel YI, John A, McHugh NJ. Antiphospholipid syndrome with proliferative vasculopathy and bowel infarction. Rheumatology (Oxford) 2000;39:108-10. [ Links ]
12. Kato N, Yukioka H, Yoshida G, Kurita S, Hirakawa K, Koh KR. Superior mesenteric vein thrombosis presented transient false positivity for lupus anticoagulant under heparin treatment. Eur J Emerg Med 2002;9:67-70. [ Links ]
13. Uthman I, Khamashta M. The abdominal manifestations of the antiphospholipid syndrome. Rheumatology 2007;46:1641-7. [ Links ]
14. Cervera R, Bucciarelli S, Plasín MA, Gómez-Puerta JA, Plaza J, Pons-Estel G, et al. Catastrophic antiphospholipid síndrome (CAPS): Descriptive analysis of a series of 280 patients from the "CAPS Registry". J Autoimmun 2009;32:240-5. [ Links ]
15. Cervera R. CAPS Registry. Lupus 2012;21:755-7. [ Links ]

