










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkMedifam
versión impresa ISSN 1131-5768
Medifam vol.12 no.1 ene. 2002
EN COLABORACIÓN CON ...
Medicina Interna
Utilización de marcadores tumorales en Atención Primaria
G. de Teresa Romero, V. Casado Vicente*, A. Jimeno Carrúez**
Residente de Medicina de Familia y Comunitaria. *Especialista en Medicina de Familia y Comunitaria.
C.S. Parquesol. Valladolid. **Especialista en Medicina Interna. Hospital Universitario. Valladolid
RESUMEN
El cáncer en España, como en el resto de países desarrollados, representa la segunda causa de muerte después de las enfermedades cardiovasculares, y la primera en cuanto a Años Potenciales de Vida Perdidos. Los Marcadores Tumorales (MT) se presentan como una herramienta útil en Atención Primaria a la hora de buscar soluciones en los tres escalones de la prevención y de buscar métodos para detectar precozmente la enfermedad, cuantificar su grado de control y seguimiento y predecir sus recidivas, si bien su efectividad y eficiencia en la práctica clínica diaria no está aún claramente establecida. A lo largo de este trabajo se revisa críticamente la evidencia existente en cuanto a las recomendaciones para el uso de los MT en el cribado, diagnóstico precoz y seguimiento de cuatro de los tumores más relevantes en nuestra práctica diaria (colon, ovario, mama y próstata), encontrando que no existe evidencia suficiente para apoyar su uso rutinario en el cribado poblacional de los mismos, aunque sí para su uso en el seguimiento y control de la respuesta al tratamiento de alguno de ellos.
Palabras clave: Marcadores Tumorales. Cribado Poblacional. Diagnóstico. Seguimiento. Atención Primaria. Medicina Basada en la Evidencia.
Clinical use of tumour markers in Primary Health Care
ABSTRACT
Cancer in Spain, as in the rest of developed countries, is the second cause of death, following cardiovascular pathologies, and the first one considering Potentially Lost Life Years. Tumour Markers (TM) show up as a useful tool in Primary Health Care when looking for solutions in the three steps of prevention and when searching for useful screening and follow-up methods, even though their effectivity and efficiency in daily clinical practice is still far from being cleared out. Along this article we critically review the existing evidence on the recommendations for the use of TM for screening, diagnosis and follow-up of four of the most relevant tumours in our daily clinical practice (colon, ovarian, breast and prostate), concluding that, on one hand, there is little evidence supporting their use for cancer screening, and, on the other hand, there seems to be enough evidence to support their use for the follow-up of some of these tumours.
Key words: Tumour Markers. Mass Screening. Diagnosis. Follow-up. Primary Health Care. Evidence Based Medicine.
ESCENARIO
El cáncer representa uno de los mayores problemas de salud pública en el mundo por el elevado número de personas a las que afecta, por su tasa de letalidad, por el número de años de vida potenciales perdidos (APVP) que genera, por la afectación de la calidad de vida que conlleva y por su repercusión familiar, sanitaria, social y económica. En España, al igual que el resto de los países desarrollados, es la segunda causa de muerte después de las enfermedades cardiovasculares. En 1995, la tasa de mortalidad por tumores malignos fue de 221,6 por 100.000 habitantes, lo que supuso el 24,3% de todas las defunciones ocurridas en ese año1. Si tenemos en cuenta los Años Potenciales de Vida Perdidos (APVP), el cáncer pasa a ocupar el primer lugar, siendo diferente según la localización de los tumores, generando mayor APVP los cánceres de pulmón y mama2. Los cánceres con mayor incidencia anual son los de pulmón, mama, colorrectal, próstata y estómago3.
El abordaje eficiente de este problema sigue siendo un reto para los profesionales de la salud, tratando éstos de buscar soluciones en los tres escalones de la prevención y de buscar métodos que sean capaces de detectar precozmente la enfermedad, cuantificar su grado de control y seguimiento y predecir recidivas. Las características que definen la Atención Primaria en nuestro sistema sanitario hacen que la aplicación de estos conocimientos cobre especial importancia. Por estos motivos, los Marcadores Tumorales podrían representar una herramienta útil y eficaz si se demuestra su validez para Atención Primaria.
DEFINICIÓN DE MARCADOR TUMORAL
Se define como marcador tumoral (MT) "toda aquella sustancia biológica o bioquímica producida o inducida por las células tumorales o por el organismo en respuesta a su presencia, vertida al torrente sanguíneo en cantidades detectables, que refleja su crecimiento y/o actividad y que permite conocer la presencia, evolución o respuesta terapéutica de un tumor maligno"4. Los MT pueden ser proteínas específicas de tumor (cromosoma Philadelphia en la leucemia mieloide crónica), proteínas no específicas de tumor o marcadores relacionados con células malignas (antígeno carcinoembrionario -CEA-, alfa-fetoproteína -AFP-, etc.), y proteínas específicas de células sobreexpresadas en células malignas (antígeno prostático específico -PSA-).
Los marcadores tumorales deberían idealmente ser útiles para la detección precoz de un cáncer, establecer el pronóstico pretratamiento (estadiaje), el seguimiento postratamiento (respuesta a la terapia), predecir las recurrencias, presentar una elevada sensibilidad (S), especificidad (E) y valor predictivo positivo (VPP) y ser órgano-específicos y tumor-específicos4,5. Las características que definen la Atención Primaria (AP) en nuestro sistema sanitario hacen que la aplicación de estos conocimientos cobre especial importancia en la detección precoz de neoplasias y en el seguimiento y predicción de sus recidivas, fundamentalmente en las neoplasias que presentan mayor prevalencia.
Los MT de los que disponemos a día de hoy no son específicos de ningún cáncer, pero pueden utilizarse tres parámetros para mejorar su especificidad y discriminar si una elevación en su concentración sérica se debe a la presencia de una enfermedad benigna o maligna6: La concentración sérica del MT: los incrementos del MT deben ser inferiores en los pacientes sin neoplasia que en los pacientes con ella. Existencia de enfermedades benignas asociadas que puedan provocar falsos positivos: deben descartarse ante toda elevación de la concentración sérica de un MT (Tabla I). Control evolutivo: si se realizan dos o tres determinaciones seriadas con un intervalo entre ellas superior a la vida media del MT se puede discernir si el origen es tumoral o no. Un incremento continuo de la concentración de un MT sugiere un origen neoplásico, mientras que una estabilización o un incremento discontinuo sugieren un origen no neoplásico.

Como sucede con todas las actividades y programas de detección precoz y/o cribado de enfermedades, antes de decidir su realización hay que asegurarse de que tanto la enfermedad como las pruebas de detección y el programa cumplen los criterios de adecuación establecidos por Wilson y Junguer (características de la enfermedad: importancia, historia natural conocida, criterios diagnósticos claros y tratamiento efectivo; características de las pruebas de detección: validez, fiabilidad, reproductibilidad y aceptabilidad; características del programa: efectividad, eficiencia y factibilidad)7.
Con todo lo presentado anteriormente, vamos a repasar el papel de los marcadores tumorales en la detección precoz y detección poblacional, en el diagnóstico, seguimiento y respuesta al tratamiento de las neoplasias más prevalentes, con mayor porcentaje de mortalidad, y en las que se ha demostrado más práctica la utilización de éstos: cáncer de próstata, ovario, colorrectal y mama.
METODOLOGÍA
--Formulación de la pregunta clínica:
¿Existe evidencia científica que apoye el uso de algún marcador tumoral en el cribado, diagnóstico, seguimiento y/o pronóstico del cáncer de próstata, ovario, colorrectal y mama en nuestra práctica clínica diaria?
--Intervenciones:
Los principales marcadores tumorales empleados en la práctica clínica habitual en pacientes con cualquiera de los cuatro tipos de cáncer referidos (Tabla II).

--Estrategia de búsqueda:
Se realizó una búsqueda electrónica desde 1966 a 2000 en las bases de datos recogidas en la tabla III seguida de una búsqueda manual en fuentes no recogidas en dichas bases o en los volúmenes de otras sí recogidas pero publicadas en fechas anteriores a su año de inclusión. Se incluyeron los artículos publicados en lengua inglesa o española. Finalmente, se seleccionaron aquellas referencias incluidas en los artículos seleccionados que se consideraron relevantes en la materia. No se revisaron fuentes de literatura gris ni se indagó en posibles sesgos de publicación. Se emplearon los filtros de calidad recomendados por Sackett et al.8 para procedimientos diagnósticos y se utilizaron las palabras claves recogidas en la tabla III.

--Criterios de selección de los artículos:
Al no existir ensayos clínicos aleatorizados y controlados y sí un elevado número de estudios primarios de otras características, decidimos finalmente incluir los trabajos que cumplieran los criterios de calidad de Greenhalgh et al.9, incluyendo así:
1. Todos los estudios secundarios realizados por expertos mediante Revisión Sistemática de la literatura científica, especialmente Guías de Práctica Clínica y revisiones de éstas realizadas, basadas en la evidencia, prestando especial atención a las que contaran entre sus expertos con médicos de Atención Primaria.
2. Todos los metaanálisis encontrados.
3. Aquellos estudios primarios encontrados o citados en las referencias de los estudios secundarios que considerásemos interesantes por su diseño y aportaciones.
Se empleó como escala de evidencia y fuerza de la recomendación la propuesta por la Canadian Task Force on the Periodic Health Examination10.
RESULTADOS Y RESUMEN DE LAS EVIDENCIAS
Escenario
Cáncer de próstata (CP)
El CP es el tumor más frecuente del sexo masculino y, tras el cáncer de pulmón, es la segunda causa de muerte por cáncer en los varones. La incidencia en España es de 22,2 por 100.000 habitantes y año y la prevalencia de 200 por 100.000 habitantes y año. En las series de autopsias su prevalencia es mayor, alcanzando el 57% en mayores de 80 años. La edad media al diagnóstico es 70 años. En el momento del diagnóstico, el 60% estarán en fases precoces, mientras que el 25-35% desarrollarán metástasis durante su evolución. La supervivencia a los 5 años es del 92-95% para la enfermedad localizada, 80-83% si hay afectación regional y 29% cuando hay extensión metastásica11.
Cáncer de ovario (CO)
El CO presenta una incidencia en España de entre el 6,3 y el 13,5 por 100.000 mujeres, con una incidencia máxima entre mujeres de 45 a 65 años, siendo la probabilidad de desarrollar un CO en mujeres de menos de 75 años de aproximadamente el 1%11. A pesar de esta baja incidencia, la letalidad (supervivencia a los 5 años de aproximadamente el 30-40%) del CO por su presentación avanzada y la inexistencia de tratamientos claramente efectivos hace que sea necesario desarrollar técnicas (analíticas y de imagen) que contribuyan de forma eficiente a su cribado y detección precoz.
Cáncer colorrectal (CCR)
El cáncer de colon es el tercer tumor maligno más frecuente a escala mundial, segundo en los países occidentales, después del cáncer de pulmón en los varones y de mama en las mujeres, representando en España en 1992 el 10,7% de los tumores malignos. Esta enfermedad fue responsable de 9.085 años perdidos de vida en 1992 en nuestro país, con una media de 13 años por individuo afectado. La incidencia en España ha ido en aumento y las tasas ajustadas por edad se sitúan en torno a 25,9 en los varones y al 20,9 en las mujeres11.
Cáncer de mama (CM)
El CM es la neoplasia más frecuente en mujeres, siendo responsable de más de un millón de casos nuevos al año en el mundo, y del 18% del total de las neoplasias en mujeres. La tasa de incidencia en los países de la Comunidad Europea varía entre un 46,3 y un 72,8 por 100.000 habitantes, siendo la de nuestro país de 47,9. Esta tasa de incidencia se incrementa con la edad, doblándose con cada década que pasa hasta la menopausia, momento en que desciende de forma importante. Aparte de la edad, otros factores de riesgo universalmente aceptados son la edad tardía del primer embarazo, la nuliparidad, la menarquia y menopausia tardía, la historia familiar de CM, la existencia de patología de mama benigna anterior, la radiación, los hábitos de vida, la distribución geográfica, la toma de anovulatorios o la terapia hormonal sustitutiva. La tasa de mortalidad en los países de la Comunidad Europea varía también entre el 23,3 y el 28,6 por 100.000 habitantes, siendo en España de 16,9 (11). En comparación con otras neoplasias, la supervivencia del CM es relativamente buena, pero la morbilidad que genera tanto la enfermedad en sí como el tratamiento es elevada, por lo que tiene especial importancia desarrollar técnicas efectivas de cribado y diagnóstico precoz.
CRIBADO POBLACIONAL O SCREENING
El objetivo de cualquier prueba de cribado es identificar pecozmente una patología sobre la que posteriormente se podrá actuar de forma claramente beneficiosa. La efectividad de un programa de cribado se debe evaluar idealmente mediante estudios aleatorizados y controlados. ¿Cumplen estos requisitos el cribado del CP, CO, CCR y CM mediante el uso de MT?
Screening de cáncer de próstata
Existen opiniones contradictorias sobre el screening de CP. Aquéllas a favor argumentan que la inexistencia de un tratamiento eficaz para los CP en estadio avanzado hace que sea especialmente importante su detección precoz para disminuir su mortalidad. Aquéllas en contra, argumentan por un lado que el screening de CP no ha demostrado reducir la morbimortalidad asociada al mismo, suponiendo además, en caso de que el resultado sea positivo, una gran tasa de efectos secundarios para un paciente que, hoy por hoy, no podrá recibir un tratamiento eficaz y, por otro, que no existe ninguna prueba diagnóstica de screening que cumpla las condiciones idóneas requeridas para ello.
--¿Existe evidencia clara de que el screening de CP disminuya la mortalidad asociada al mismo?
No se ha publicado aún ningún estudio con el diseño adecuado para responder a esta pregunta. Hemos encontrado sólo tres estudios que cumplan estas características e intenten o hayan intentado aclarar el balance riesgo/beneficio del screening de CP. Dos de ellos están actualmente en desarrollo, uno en Europa y el otro en EE.UU., y sus resultados no estarán listos hasta, al menos, el año 2006-200812,13. El estudio europeo incluye 190.000 varones de entre 50 y 70 años de cinco regiones distintas de Europa, esperándose sus resultados para el año 2008. Su objetivo es estudiar varones asintomáticos con determinaciones iniciales de PSA, aleatorizándolos posteriormente a un grupo de screening o a un control en el que se realizará tacto rectal y ecografía transrectal. El estudio americano incluye 79.000 varones de entre 55 y 74 años, estudiados con tacto rectal, antígeno prostático específico (PSA) y ecografía transrectal cuando se precise, y sus resultados se esperan para el año 2006. El tercer estudio, el primero de estas características que se realizó sobre screening de CP, se llevó a cabo en Canadá en 1999. Labrie et al.14 aleatorizaron 46.193 varones a un grupo de intervención (screening) o a un grupo control (no screening). Sólo 5 de los 8.137 sujetos del grupo de intervención murieron, frente a los 137 de los 38.056 del grupo control. Las tasas de mortalidad asociadas a CP fueron de 48,7 versus 15 por 100.000 varones y por año en el grupo control e intervención, respectivamente, lo que supone una odds-ratio de 3,25 a favor del screening (p<0,01). No obstante, el estudio estuvo sesgado desde el momento en que la distribución de los sujetos fue desigual, pues sólo se realizó screening al 23% de los sujetos aleatorizados al grupo de intervención15,16.
Por lo tanto, es necesario esperar a los resultados de los dos importantes estudios que en estos momentos se están desarrollando para poder determinar con seguridad los riesgos/beneficios del screening de CP y si éste reduce la incidencia de mortalidad asociada al mismo.
1. ¿Qué es el PSA y qué aporta al screening de CP? ¿Qué otros métodos existen para aumentar su sensibilidad y/o especificidad?
El PSA es una glicoproteína de cadena simple de 33 Kda. de peso molecular que fue indentificada por primera vez en el líquido seminal por Hara et al. en 197117 y aislada en el tejido prostático por Wang et al. en 197918. Está producida por el epitelio secretor prostático y se encuentra normalmente en concentraciones de 0,1-4,0 ng/ml en plasma. No obstante, no es una proteína específica de malignidad ni tampoco específica de órgano19, como se recoge en muchos trabajos de revisión, pues se ha demostrado con métodos sensibles que existe también en células de las glándulas periuretrales20, en mujeres21, en otras patologías prostáticas (Tabla I), en la eyaculación22, en el ejercicio físico, las cistoscopias o las biopsias prostáticas23.
Existe gran controversia en la literatura científica a la hora de establecer los valores diagnósticos del PSA como test de screening (sensibilidad, especificidad y valor predictivo positivo), pues están íntimamente relacionados con las características de cada estudio (diseño, número de sujetos, métodos estadísticos empleados, etc.). Además, en todos los estudios encontrados se decide la realización de biopsia (prueba más frecuentemente escogida como gold standard) en el caso de que se halle alguna prueba alterada, no realizándose cuando éstas son normales, generándose un sesgo importante en los resultados. En estas condiciones, tan sólo el VPP podría ser calculado con alguna confianza, pero no la sensibilidad ni la especificidad. Otro de los problemas encontrados a la hora de calcular estos valores es la gran heterogeneidad existente con la biopsia glandular. Las distintas condiciones en que ésta se ha realizado en los estudios publicados (material empleado, zona biopsiada, etc.) y los niveles de PSA considerados justifican la gran horquilla de valores dada por los diferentes autores sobre la sensibilidad (S), especificidad (E) y valor predictivo positivo (VPP) del PSA. Se han publicado múltiples estudios con resultados muy dispares, encontrándose VPP que oscilan entre 10-80% (C13), S entre 29-80%23, 25 y E entre 60-91%26.27.
Tradicionalmente, se ha establecido el punto de corte ideal para diferenciar CP de HBP en un valor de PSA de 4,0 ng/ml. Sin embargo, en otros estudios con varones mayores de 50 años, 136 de los 319 que presentaron CP (43%) mostraron valores de PSA<4,0 ng/ml, mientras que 148 de los 597 (25%) sin CP presentaron valores de PSA>4,0 ng/ml28,29. En otro estudio, 332 varones asintomáticos con tacto rectal (TR) y PSA normales fueron sometidos a biopsia, encontrándose CP en 73 (22%) de ellos, todos localizados30.
Con el objetivo de aumentar esta especificidad (objetivo principal en varones >60 años) sin disminuir la sensibilidad (objetivo principal en varones <60 años) se han propuesto varias alternativas:
--Cociente PSA libre/PSA total:
Steinmam (1991) fue el primer autor en observar que los pacientes con hiperplasia benigna de próstata (HBP) presentaban valores superiores a los pacientes con CP31. Hemos encontrado al menos ocho estudios a favor y tres en contra de este parámetro, cuya descripción sobrepasa el objetivo de este trabajo.
El punto de corte ideal sigue siendo desconocido. Se han propuesto valores tan dispares como 14-28% para mantener una sensibilidad superior a 90% y especificidades de entre 19-64%, lo cual implicaría que, al menos, el 36% de las biopsias realizadas no serían de CP32.
Por ello, el cociente PSA libre/PSA total puede aportar información importante cuando el PSA total sérico está en valores entre 2,5-10 ng/ml, pero por encima de éstas aporta información poco valiosa. Se necesitan estudios prospectivos multicéntricos y correctamente diseñados para establecer la validez definitiva que este parámetro tiene en la diferenciación de CP e HBP.
--Densidad de PSA (DPSA) (PSA relativo al tamaño prostático):
Benson et al. en 199233 demostraron que las células benignas tienen una capacidad máxima de síntesis de PSA y una menor variabilidad de éste, por lo que valores anormalmente altos han de hacernos sospechar la existencia de cáncer. Corroborada por algunos autores34,35, esta hipótesis sigue en entredicho36,37. No sin polémica, el valor más aceptado para diferenciar malignidad de benignidad es 0,1538.
Hoy por hoy, la DPSA no ha sido adoptada sistemáticamente por ninguna Sociedad de Expertos como método habitual de screening.
--Velocidad de PSA:
Carter et al. en 199239 descubrieron que la velocidad de incremento de PSA era significativamente superior en CP que en HBP. Observaron una velocidad de incremento mayor de 0,75 ng/ml por año en pacientes con CP durante los casi 9 años que pasaban hasta que el tumor se hacía clínicamente importante. Las críticas a la aplicación diaria de este parámetro surgen por la elevada variabilidad biológica del PSA y por la existencia de muy distintos métodos de laboratorio para su determinación40,41.
Es necesario realizar varias determinaciones seriadas de PSA para considerar la elevación significativa, no quedando claro los intervalos con que las mismas deben realizarse42, 44. Como los anteriores parámetros, es de utilidad para complementar la información aportada por el PSA total sérico, especialmente cuando éste está dentro de los límites de la normalidad.
--PSA ajustado a la edad:
El primer autor en hacer referencia a la existencia de correlación entre la edad y los valores de PSA fue Oesterling45. Demostraron en un grupo de 471 varones sanos de entre 40 y 79 años, que existía correlación entre el PSA sérico y la edad (r=0,43; p<0,0001) y entre el PSA sérico y el volumen prostático (r=0,55; p<0,0001), estando éste relacionado con la edad (r=0,43; p<0,0001) y débilmente entre la edad y la densidad de PSA (r=0,25; p<0,001). Encontraron que la concentración de PSA sérico aumenta una media de 0,04 ng/ml por año, proponiendo los siguientes valores ajustados por rango de edad (IC 95%) para aumentar la sensibilidad en jóvenes y la especificidad en ancianos: de 40-49 años: 0,0-2,5 ng/ml; de 50-59 años: 0,0-3,5 ng/ml; de 60-69 años: 0,0-4,5 ng/ml y de 70-79 años: 0,0-6,5 ng/ml. Dos años más tarde46 presentaron los resultados de su estudio tomando como referencia estos valores de PSA ajustados por edad, encontrando que aplicándolos se hubieran evitado el 5,5% de las 1.686 biopsias perdiendo tan sólo el 0,6% de los CP, la mayoría de los cuales serían clínicamente poco importantes, resultados confirmados por Partin et al. en 199647. No obstante, otros autores no han corroborado estos datos48,49.
Debido a lo contradictorio de los datos, se necesitan otros estudios para concluir poder recomendar su uso rutinario.
2. ¿Es el PSA capaz de diferenciar entre los CP clínicamente importantes y los que no lo son?:
No existe una definición universalmente aceptada de lo que significa 'CP clínicamente importante'. Se ha intentado definir midiendo el volumen prostático, el estadiaje y el grado de diferenciación, la existencia de afectación de los bordes quirúrgicos, la clasificación de Gleason, etc. Una gran parte de los CP detectados mediante PSA son clínicamente importantes50-52, y esta capacidad de diferenciación aumenta cuando se combinan distintos métodos (PSA, estadio clínico y clasificación de Gleason)53, aunque también existe evidencia de que el PSA no detecta muchos de los tumores de pequeño volumen, que son los más prevalentes, pues cerca de un tercio de los tumores detectados mediante PSA reúnen características de no localizados (extracapsulares, baja diferenciación, diseminación a distancia, etc.)30.
Se necesitan, por tanto, más estudios prospectivos adecuadamente diseñados para resolver esta cuestión.
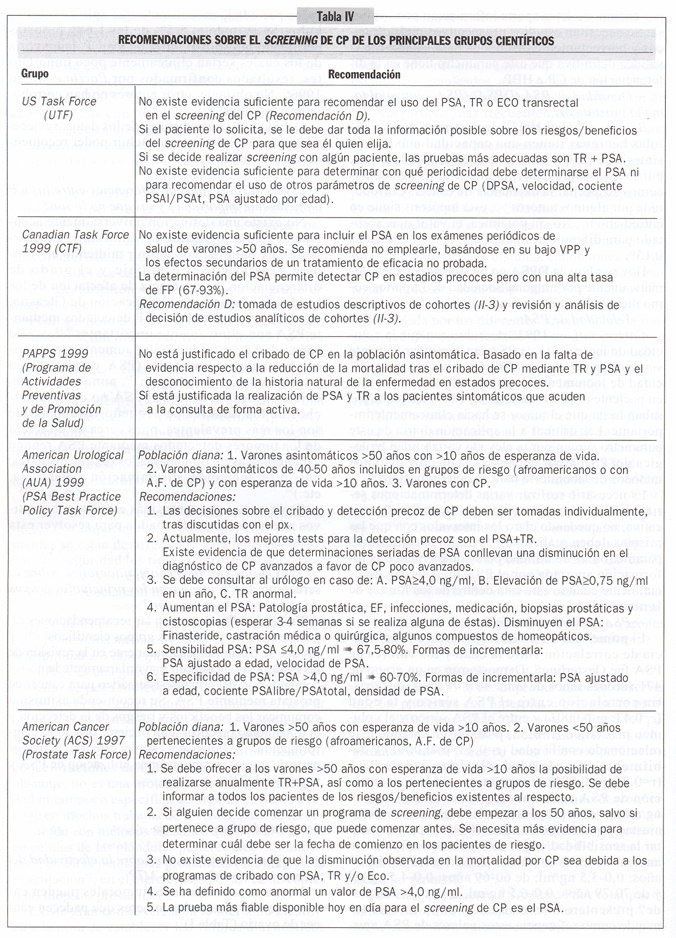
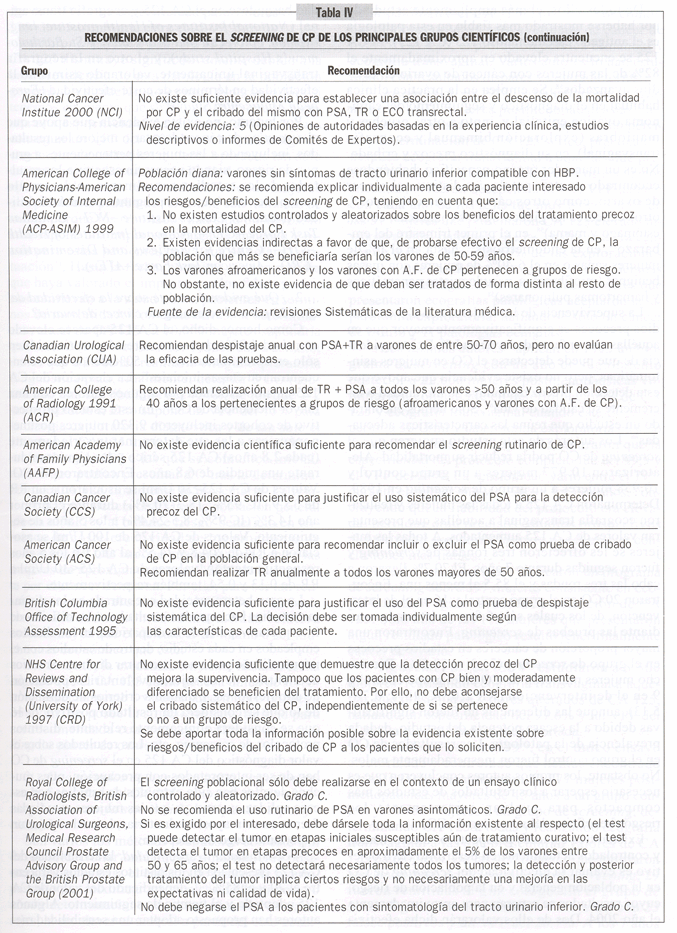
3. ¿Cuáles son las recomendaciones sobre el screening de CP que hacen los principales grupos de trabajo sobre prevención?:
En la tabla IV se recogen las recomendaciones realizadas por los principales grupos científicos23, 54-62 y los que se basan fundamentalmente en la revisión de las evidencias establecen mayoritariamente la no-recomendación del cribado sistemático para cáncer de próstata mediante PSA. Se recomienda asimismo comunicar los beneficios y riesgos de la detección y el tratamiento precoces a un paciente que solicita información. De llevarse a cabo el cribado, la mayor parte de grupos recomiendan la utilización de PSA y tacto rectal (TR).
Screening de cáncer de ovario
1. ¿Qué evidencia existe sobre la efectividad del 'screening' de CO usando MT?
Numerosos marcadores tumorales pueden encontrase elevados en las mujeres que padecen cáncer de ovario (Tabla I).
De todos ellos, el más ampliamente estudiado por haberse mostrado más fiable en esta patología es el antígeno carcinomatoso 125 (CA 125). El CA 125 se encuentra elevado en aproximadamente el 82% de las mujeres con cáncer de ovario en estadios avanzados58. Se emplea en la práctica clínica habitual en el diagnóstico y seguimiento del carcinoma ovárico epitelial y, junto a otras pruebas y maniobras (exploración bimanual y ecografía transvaginal), en su diagnóstico precoz y cribado. No es un marcador órgano-específico, habiéndose encontrado en otras patologías distintas al cáncer de ovario, como otros cánceres ginecológicos63, otros cánceres no ginecológicos (páncreas, colon, estómago y mama)58, en el primer trimestre del embarazo64, en la endometriosis65, hasta en el 1% de mujeres sanas o en el 6-40% de mujeres con masas benignas (miofibromas, pseudoquistes pancreáticos y hamartomas pulmonares)58.
La supervivencia de las mujeres con CO en estadios precoces es significativamente mayor que en aquéllas con enfermedad avanzada66. Existe evidencia de que puede detectarse el CO en mujeres asintomáticas, pero no existe evidencia que apoye que esta detección precoz disminuya su mortalidad o incremente su calidad de vida67. Sólo hemos encontrado un estudio que reúna las características adecuadas68. Los autores de este estudio sugieren que el screening de CO podría reducir su mortalidad. Aleatorizaron 10.977 mujeres a un grupo control y 10.958 mujeres a un grupo de screening en 1989. Determinaron CA 125 a todas las mujeres y realizaron ecografía transvaginal a aquéllas que presentaran valores de CA 125 aumentados. A todas las mujeres se les ofrecieron tres rondas de screening y fueron seguidas durante 7 años. El 70,7% llevaron a cabo las tres rondas y el 85,5 al menos una. Encontraron 20 CO en el grupo control y 16 en el de intervención, de los cuales sólo 6 fueron detectados mediante las pruebas de screening. Encontraron una mayor proporción de cánceres en estadios precoces en el grupo de screening (31,3 vs 10,0%). Dieciocho mujeres murieron de CO en el grupo control y 9 en el de intervención (RR 2,0; IC 95%: 0,78-5,13), aunque las diferencias no fueron significativas debido a la escasa potencia del estudio, dada la prevalencia de la patología. Los resultados de CO en el grupo control fueron inesperadamente malos. No obstante, los propios autores concluyeron que es necesario esperar a los resultados de estudios más compactos para evaluar correctamente los riesgos/beneficios del screening de CO.
Existen tres estudios prospectivos aleatorizados y controlados actualmente en desarrollo cuyo objetivo es evaluar la efectividad del screening de CO en la población general y en la población de riesgo, cuyos resultados se esperan para aproximadamente el año 2004. Dos de ellos valorarán dicha efectividad basándose en el CA 125 y ecografía transvaginal (National Institutes of Health prostate, lung, ovarian, colorectal and ovary study y St. Bartholomew´s Hospital study), y el otro en la ecografía transvaginal únicamente, valorando asimismo la efectividad en términos de coste-efectividad (European Multicentre Study)69,70.
Por lo tanto, no existe evidencia que apoye que el screening de cáncer de ovario mejore los resultados, incluyendo a las mujeres pertenecientes a grupos de riesgo, siendo necesario esperar a los resultados de los estudios actualmente en desarrollo para poder resolver esta pregunta con más fiabilidad (National Cancer Institute -NCI-, Canadian Task Force -CTF-, National Institutes of Health -NIH-, Centre for Reviews and Disemination -CRD-, American Task Force -ATF-).
2. ¿Qué evidencia existe sobre la efectividad del CA 125 para el screening de cáncer de ovario?
Como hemos dicho, el CA 125 aparece elevado en más del 80% de los tumores de ovario, si bien sólo en aproximadamente el 50% de los que se encuentran en etapas iniciales58. La elevación del CA 125 ha sido asociada por algunos autores con una mayor incidencia de CO71. En este estudio prospectivo de cohortes incluyeron 9.320 mujeres postmenopáusicas a las que determinaron seriadamente (cada 2,8 años) CA 125 sérico y las siguieron durante una media de 6,8 años. Encontraron 49 CO. Valores de CA 125>=30 U/ml se asociaron a un RR de 35,9 (IC 95%: 18,3-70,4%) durante el primer año 14,3% (IC 95%: 8,5-24,4%) a los 5 años de seguimiento. Valores de CA 125 de 100 U/ml se asociaron a RR de 204,8 y 74,5% al año y a los 5 años respectivamente y valores de CA 125<30 U/ml a RR de 0,13 y 0,54, también respectivamente.
La gran heterogeneidad encontrada en los distintos estudios hace que los resultados sean difíciles de interpretar. Aparte de la disparidad en los diseños empleados en cada estudio, dentro de estudios con el mismo diseño podemos encontrar distintos criterios de inclusión de los sujetos (voluntarios, población de alto riesgo, etc.), distintos criterios de definición de lo que se considera un resultado positivo, de lo que se considera clínicamente relevante, distintos protocolos de screening, etc. Los resultados sobre el valor diagnóstico del CA 125 en el screening de CO han de ser interpretados con precaución, pues muchos de ellos han sido extraídos de estudios con escasa validez externa, ya que las mujeres estudiadas fueron en su mayoría voluntarias, lo cual supone un importante sesgo de selección:
--Sensibilidad y especificidad: la sensibilidad del CA 125 presentada en los distintos estudios varía entre el 20 y el 85%72,73, dependiendo del valor de CA 125 adoptado y el tiempo de seguimiento. Algunos autores han propuesto adoptar una sensibilidad media del 75%73. La especificidad para valores de CA 125>30-35 U/ml varía entre el 95-99,9%72, 74.
--Valor predictivo positivo (VPP): el VPP del CA 125 disminuye significativamente debido a la baja prevalencia de esta patología en la población general. La situación no es muy esperanzadora si tenemos en cuenta que se ha calculado que se necesita una especificidad de al menos el 99,6% para conseguir un VPP del 10%, lo cual seguiría significando que de cada diez mujeres operadas tan sólo una presentaría la enfermedad77. En la práctica real, aproximadamente entre el 3-12% de las mujeres que pasan una prueba de screening son llamadas de nuevo porque los resultados han sido positivos, lo cual supone una importante carga de ansiedad y estrés para estas pacientes hasta que se aclara la situación73. Hasta la fecha, no existe ningún estudio que haya valorado el impacto que los falsos positivos conllevan a la salud de las pacientes en términos de morbilidad. Con estas cifras, la determinación anual de CA 125 en 10.000 mujeres supondría que unas 300 volverían a ser requeridas a estudio, de las que a 20 se les realizaría cirugía exploratoria diagnóstica para, finalmente, detectar 3 CO (asumiendo una sensibilidad del 75%), de los que 1-2 serán estadios I. Esto supone un VPP del 15% para la cirugía exploratoria diagnóstica y de tan sólo el 1% para el CA 125 inicial. Es decir, que tan sólo 1 mujer de cada 100 a las que se comunicó inicialmente que podían presentar CO tras la determinación de CA 125 finalmente lo presentaría. La relativa baja prevalencia de CO en la población general afecta al VPP y al balance riesgo-beneficio en términos de coste-efectividad de los programas de screening. El CO supone una mortalidad entre las mujeres de aproximadamente el 30% de la supuesta por el cáncer de mama. Por ello, para ser tan rentable como éste, el screening de CO debería resultar en una reducción de la mortalidad relativa mucho mayor que el de cáncer de mama o ser mucho más económico que éste.
Por lo tanto, la evidencia existente hasta la fecha sugiere que el CA 125 como prueba única de screening, especialmente a valores superiores o iguales a 35 U/ml, no posee la suficiente sensibilidad para ser recomendada como prueba rutinaria de screening de cáncer de ovario.
3. ¿Qué métodos se han propuesto para aumentar la fiabilidad del CA 125 como prueba de screening (aumentar la sensibilidad sin perder especificidad)?
Son varios los métodos propuestos con este fin: desde seleccionar únicamente a las mujeres postmenopáusicas para el cribado de cáncer de ovario, pasando por modificar las técnicas de laboratorio o añadir determinaciones de otros marcadores tumorales, hasta elevar el umbral a partir del cual considerar patológico el CA 12558, pasando por combinar distintas pruebas para el cribado de cáncer de ovario: CA 125, exploración pélvica bimanual y ecografía transvaginal. Varios autores lo han estudiado, con resultados dispares y contradictorios:
--Jacobs et al. en 198875 estudiaron prospectivamente a 1.010 mujeres postmenopáusicas asintomáticas mayores de 45 años con exploración pélvica y determinación de CA 125 sérico, practicando después ecografía transvaginal a aquéllas que presentaran algún resultado patológico. Los resultados encontrados fueron decepcionantes: el VPP hallado con cada uno de los tests fue muy bajo. Sólo 3 de las 31 mujeres que presentaron CA 125 elevado tuvieron alguna patología, de las cuales una era cáncer de ovario. Más del 50% de las exploraciones pélvicas que resultaron patológicas fueron debidas a patología benigna. Doce de las trece mujeres que presentaron ecografías patológicas aceptaron la laparotomía exploratoria, hallándose sólo un cáncer de ovario y 11 patologías benignas.
--Los mismos autores81 aplican en 1993 un programa de screening con un año de seguimiento posterior a 22.000 mujeres postmenopáusicas voluntarias, consistente en determinaciones de CA 125 y ecografía transvaginal cuando éste fuese superior a 30 U/ml y laparotomía exploratoria cuando la ecografía fuese patológica. Encontraron que la especificidad del protocolo completo fue del 99,9 y el VPP del 27%. La sensibilidad varió entre el 79% durante el primer año y el 58% a los dos años, concluyendo que la fiabilidad del protocolo era elevada, pero que eran precisos estudios prospectivos aleatorizados y controlados para poder extraer conclusiones más fiables.
--Karlan et al. en 199376 aplicaron un programa de screening sobre 597 mujeres consistente en ecografía transvaginal asociada a detección de CA 125. Sólo una de las 115 mujeres que presentaron algún resultado patológico resultó tener finalmente cáncer de ovario.
--Nguyen et al. en 199477 estudiaron a 5.500 mujeres, realizando ecografía transvaginal a aquéllas que presentaran valores alterados de CA 125, hallando un especificidad del 97,6% y un VPP del 50%. La sensibilidad no fue reportada.
--Jacobs et al. en 199968, en el único ensayo aleatorizado y controlado que hemos encontrado hasta la fecha, aleatorizaron 10.977 mujeres postmenopáusicas mayores de 45 años a un grupo control y 10.958 mujeres a un grupo de screening, determinando CA 125 a todas y realizando ecografía transvaginal a las que presentaran valores de CA 125 patológicos. De las 468 mujeres que mostraron elevación del CA 125 en el grupo de screening, 29 fueron remitidas al ginecólogo. En el grupo de screening se detectó cáncer en 6 mujeres, con 23 falsos positivos y un VPP del 20,7%. A los 7 años de seguimiento se detectaron 10 casos de CO en el grupo de screening y 20 en el grupo control. La supervivencia media en el grupo de screening fue de 72,9 meses, siendo la del grupo control de 41,8 meses. No se encontraron diferencias significativas en la mortalidad asociada a CO entre los dos grupos. No obstante, las conclusiones de este estudio han de interpretarse con precaución, pues el objetivo primario del estudio no era evaluar el impacto del screening en la mortalidad. Los autores concluyeron el trabajo diciendo que se necesitan más estudios con un objetivo inicial de valorar el impacto del screening en el descenso de la mortalidad asociada a CO para aclarar dicha polémica.
Los estudios basados en modelos de decisión que han estudiado la efectividad del screening del cáncer de ovario con ecografía transvaginal y determinación de CA 125 han concluido que, a pesar de la elevada especificidad estimada con la combinación de ambos métodos (99,95%), el incremento de la expectativa de vida observado es menor de un tercio de día por mujer menor de 65 años y menor de tres cuartos de día en mujeres mayores de 65 años, concluyendo que el screening masivo de cáncer de ovario no puede ser recomendado como método eficiente de política sanitaria78,79.
Lo contradictorio de los resultados hace que sea necesario esperar a las conclusiones de los estudios prospectivos actualmente en desarrollo para poder concluir con mayor evidencia los riesgos/beneficios del screening de CO mediante la asociación de las pruebas referidas.

4. ¿Qué evidencia existe sobre la efectividad del screening de CO en población de riesgo?
Aproximadamente el 5-10% de los cánceres de ovario son familiares, habiéndose identificado 3 patrones hereditarios distintos: cáncer de ovario aislado, cáncer de ovario asociado a cáncer de mama y cáncer de ovario asociado a cáncer de colon. El mayor riesgo para padecer cáncer de ovario parece ser que tiene antecedentes familiares de primer grado, siendo máximo en mujeres con antecedentes de dos o más familiares de primer grado con cáncer de ovario80.
En estos grupos de alto riesgo, determinadas mutaciones genéticas parecen conllevar un mayor riesgo de desarrollar CO. La mayoría de las familias afectadas con el síndrome del cáncer de ovario y mama tienen en común la mutación del gen BRCA 1 del cromosoma 17q2181,82, que también ha sido hallada en el 5% (IC 95%: 3-8%) de las mujeres diagnosticadas de CO aislado antes de los 70 años68, habiendo quedado demostrado que las mujeres que presentan esta mutación tienen un mayor riesgo de desarrollar CO a lo largo de su vida83,84. Una proporción algo menor de mujeres con el síndrome del cáncer de ovario y mama presentan una mutación del gen BRCA 2 situado en el cromosoma 13q1285. Por todo ello, la identificación de éstas podría suponer en un futuro un importante avance en la detección precoz de esta patología en estas mujeres.
La mayor prevalencia de CO encontrada en esta población de riesgo supone que las pruebas de screening en las mismas vayan a ser más sensibles y a tener mayor VPP. Además, este grupo poblacional será más afín a participar en programas de screening, resultando en una modificación de los resultados que habrá que tener en cuenta a la hora de interpretarlos para evitar cometer errores. Sin embargo, no existe hasta la fecha evidencia alguna definitiva que haya demostrado claramente un balance positivo en riesgos-beneficios y coste-efectividad en el screening de cáncer de ovario en la población de alto riesgo67,73,78.
5. ¿Qué recomendaciones establecen los Grupos de Trabajo sobre el screening de CO?
Las recomendaciones de los grupos de trabajo se recogen en la tabla VI.11, 57-59, 66, 86-91.


Existe unanimidad en cuanto a no recomendar el screening de ovario mediante MT, ecografía transvaginal y/o exploración pélvica en mujeres asintomáticas. Aunque el National Institute of Health Consensus Statement on Ovarian Cancer recomienda determinaciones anuales de CA 125, exploración y ecografía transvaginal hasta los 35 años, edad en la que recomiendan ooforectomía bilateral profiláctica en las mujeres pertenecientes a grupos de riesgo, todos los grupos restantes establecen que no existe evidencia suficiente para hacer recomendaciones a favor o en contra (ATF, CTF, CRD, NCI y Programa de Actividades Preventivas y de Promoción de la Salud -PAPPS-).
Screening de cáncer colorrectal y cáncer de mama
Todos los grupos de trabajo de CCR y CM coinciden en que, actualmente, de los múltiples MT conocidos, ninguno ha demostrado ser eficiente en su detección precoz debido a su baja sensibilidad y especificidad en etapas iniciales ( VI).
SEGUIMIENTO, RESPUESTA AL TRATAMIENTO Y PRONÓSTICO
Existen decenas de marcadores tumorales que están siendo estudiados para ver si pueden ser utilizados en el diagnóstico, seguimiento y pronóstico de los distintos tumores (II). Hasta la fecha de hoy, no existe evidencia suficiente para recomendar su uso rutinario en ninguna de estas etapas. Aparte de estar aún en fase de estudio, su aplicación en Atención Primaria hoy por hoy es mínima, por lo que su revisión excede el objetivo de este trabajo. Nos limitaremos, pues, a citar algunas de las características de los que parecen ser los más importantes.
Cáncer de próstata
1. ¿Existe evidencia de que el PSA ayude en el manejo y seguimiento del CP?
Aparte de su empleo en el cribado y diagnóstico precoz del CP, el PSA se usa de forma habitual en el manejo del mismo, como indicador de respuesta al tratamiento o de recidiva y/o progresión. A medida que el PSA se ha ido convirtiendo en el marcador tumoral más aceptado en el CP, la mayoría de los estudios se han basado en sus variaciones para definir lo que puede considerarse buena y mala respuesta al tratamiento. Muchos autores han considerado 'buenos respondedores' a aquéllos que muestran descensos en los valores de PSA de entre el 50 y el 80% con respecto a los valores pretratamiento. Suele considerarse 'respondedor total' a aquél que presenta valores no detectables de PSA tras prostatectomía radical o a los que presentan descensos del PSA hasta valores normales (habitualmente menores de 4 ng/ml) independientemente del tratamiento recibido. Lo que sí parece estar claro es que el valor del PSA pretratamiento es un predictor independiente de respuesta a todas las formas de tratamiento. Los pacientes que presentan valores inferiores a 10 ng/ml tienen más posibilidades de responder mejor al tratamiento local91.
--PSA en el seguimiento post-prostatectomía radical (PR):
Teóricamente, las cifras de PSA deberían hacerse indetectables a las 3-6 semanas una vez realizada la prostatectomía radical. La mayoría de los algoritmos recomiendan una determinación de PSA a los 3 meses de la cirugía. Encontrar valores detectables de PSA va a favor de que exista enfermedad residual o recurrencia92. Algunos autores han establecido un valor de PSA de 1,0 ng/ml para indicar al existencia de recurrencia (local o a distancia)93. Más recientemente, el empleo de métodos ultrasensibles de detección de PSA ha permitido establecer en 30 pg/ml el límite para detectar existencia de enfermedad residual tras prostatectomía radical21, quedando descrito qué incrementos de PSA postoperatorios de entre 0,001-0,1 ng/ml son indicadores de mal pronóstico94.
Catalona y Smith en 1994 encontraron como factores pronósticos de recurrencia tras cirugía radical de la próstata: el estadiaje tumoral, la presencia o ausencia de bordes libres quirúrgicos, la diferenciación tumoral o la concentración de PSA pretratamiento95. Este último factor ha sido estudiado por distintos autores, viéndose que con PSA normales antes de la intervención la probabilidad de no-recidiva a los 5 y 10 años oscila entre 6996 y el 95%95, mientras que la probabilidad de no-recidiva con PSA preoperatorios >10 ng/ml oscila entre 4796 y el 71%95.
Otros índice empleados como factores pronósticos han sido 'el tiempo de doblaje de concentración de PSA' (una elevación mantenida del PSA tras PR indica recidiva local o a distancia, tendiendo el 'tiempo de doblaje del PSA' y la 'velocidad de PSA' a ser más lentos y lineales en las recidivas locales, mientras que las recidivas a distancia suelen presentar valores más altos de PSA en espacios más cortos de tiempo)97 y la expresión de la proteína del oncogen Bcl-2 y de la p5398.
--PSA en el seguimiento postradiectomía:
El papel de la radioterapia en el tratamiento del CP es claro. Cuando hay afectación ósea, la radioterapia alivia el dolor y disminuye el riesgo de fractura. Al igual que en la cirugía, el PSA es actualmente el principal marcador de seguimiento tras radiectomía prostática99. A diferencia de la primera, tras la radiectomía el PSA tarda bastante en alcanzar valores normales o, incluso, nunca llega a normalizarse97. El valor de referencia para asegurar que no existe enfermedad bioquímica está aún por determinar, aunque valores mínimos menores de 1,0 ng/ml se asocian generalmente a tasas de supervivencia a los 5 años muy elevadas99,100. La American Society of Therapeutic Radiology and Oncology (ASTRO) define 'fracaso terapéutico' como la determinación de 3 valores consecutivos de PSA incrementados101. El factor pronóstico más importante de éxito terapéutico es el valor más bajo de PSA tras la radioterapia, de forma que valores inferiores a 0,5 ng/ml se asocian a supervivencias a los 5 años superiores al 93%, mientras que valores superiores a 1,0 ng/ml lo hacen a supervivencias de sólo el 26%100.
No obstante, se necesitan nuevos datos para asegurar con certeza en qué medida existe asociación entre los valores del PSA y la respuesta al tratamiento radioterápico del CP.
Otros marcadores bioquímicos actualmente en estudio son la fosfatasa alcalina, la interleukina 6, el factor de crecimiento de fibroblastos, el factor de crecimiento endotelial, los marcadores neuroendocrinos, la endotelina 1, la osteocalcina y la prolactina102.
--PSA en el seguimiento postratamiento hormonal:
El tratamiento hormonal es el de elección en CP localmente avanzado o con afectación a distancia. No hemos encontrado ningún estudio aleatorizado y controlado sobre el seguimiento monitorizado de PSA de pacientes con CP en tratamiento hormonal, por lo que no existe consenso sobre los valores ideales de PSA y su periodicidad en el seguimiento tras tratamiento hormonal. Los diferentes agentes terapéuticos empleados con sus diferentes mecanismos de acción, las diferencias intersubjetivas, y otras muchas variables dificultan la unificación de criterios. No obstante, en los estudios publicados, el PSA parece seguir siendo el mejor marcador de respuesta y predictor de supervivencia. Carducci et al., en una revisión realizada en 1999, concluyen que descensos en los valores de PSA del 50% en la 4-8 semanas posteriores al inicio del tratamiento implican una buena respuesta al mismo102. Descensos en el PSA se asocian a mejoras en la calidad de vida y en la afectación de tejidos blandos por parte del tumor, como han demostrado distintos autores103,104.
2. ¿Qué recomendaciones establecen los Grupos de Trabajo sobre el PSA en el manejo y seguimiento del CP?
Las recomendaciones sobre el PSA en el manejo y seguimiento de CP se exponen en la tabla VI. Los grupos de trabajo recomiendan la monitorización periódica de PSA y TR en la prevención de recurrencias. La ASTRO recomienda determinaciones cada 3-6 meses para detectar posibles recurrencias.
Cáncer de ovario
Actualmente, el único MT que ha sido validado en el seguimiento del CO es el CA 125. Las variaciones en las determinaciones de este marcador aportan información relevante en cuanto a la respuesta al tratamiento quirúrgico y quimioterápico y aparición de recidivas, pero su papel en el diagnóstico y pronóstico de CO no está aún del todo esclarecido105-108. Algunos autores han asociado descensos rápidos de los valores de CA 125 tras el tratamiento quimioterápico a mejores pronósticos en pacientes con CO en estadio avanzado. Está descrito que los valores de CA 125 preoperatorios pueden discriminar entre masas malignas y benignas en las mujeres postmenopáusicas. En mujeres premenopáusicas, la especificidad es muy baja. De Brujin et al. encontraron que los valores elevados de CA 125 preoperatorio en CO estadio I se asocian a un riesgo de mortalidad asociada a la enfermedad seis veces mayor que los valores normales109.
El resto de MT para monitorizar el seguimiento del CO está aún en estudio. Ante cifras preoperatorias elevadas de CA 125 la determinación de otros marcadores tumorales no aporta información de interés. Sin embargo, ante cifras normales, esta determinación sí puede ser de utilidad110. Algunos autores han propuesto como medida para mejorar la especificidad del CA 125 su asociación al Antígeno Sérico Asociado a Cáncer (CASA), considerándola especialmente útil en los casos de tumores que no secretan cantidades importantes de CA 125111.
Por lo tanto, podemos concluir que las variaciones en las determinaciones de CA125 aportan información relevante en cuanto a la respuesta al tratamiento quirúrgico y quimioterápico y aparición de recidivas, pero su papel en el diagnóstico y pronóstico de CO no está aún del todo esclarecido.
Cáncer colorrectal
1. ¿Qué es el antígeno carcinoembrionario (CEA) y qué aporta al seguimiento del CCR?
El CEA es una proteína oncofetal asociada a cáncer gastrointestinal que se encuentra elevada frecuentemente en el CCR. Fue descubierto por primera vez en 1965 por Gold y Freedman112. Puede también encontrase elevado en otras enfermedades malignas y benignas (Tabla I) o incluso en pacientes sin enfermedad aparente. En las enfermedades malignas, el CEA parece elevarse por distintos motivos: aumento del número de células productoras de CEA, incremento de la capacidad de síntesis de CEA en las células malignas y/o disminución de la capacidad de eliminación de esta sustancia. Su aclaramiento se hace vía hepática, razón que justifica el hecho de que las mayores concentraciones de CEA puedan observarse cuando existen metástasis en este órgano113.
Cifras elevadas en el CEA preoperatorio se han considerado indicadoras de peor pronóstico, especialmente aquéllas que no descienden tras la cirugía114 y se asocian a mayor incidencia de recurrencias tras resección quirúrgica del tumor. Slentz et at. encontraron en 1994 que el peor pronóstico lo tenían aquéllos cuyas cifras de CEA se encontraban elevadas antes y después del tratamiento (66% supervivencia a los 5 años), en comparación con aquéllos que mostraron cifras elevadas antes pero no después del mismo (supervivencia 88% a los 5 años) y aquéllos que mostraron cifras normales tanto antes como después del tratamiento (supervivencia 93% a los 5 años)115. El grado de la elevación del CEA parece correlacionarse con el estadio del tumor (Dukes), de tal forma que valores superiores a 20 ng/ml son indicativos de enfermedad avanzada de forma casi inapelable116. Clásicamente, Wanebo et al. establecieron una asociación entre la elevación del CEA por encima de 5 ng/ml y el estadiaje del tumor: 4% Dukes A, 26% Dukes B, 44% Dukes C, 65% Dukes D y 72% en pacientes con enfermedad recurrente o metastásica113.
2. ¿Qué otros MT pueden actuar como factores pronóstico?
Existe cada vez mayor evidencia de que ciertas mutaciones de oncogenes específicos (Ki-ras), deleciones o inactivaciones de genes supresores del tumor (p53, genes reparadores del DNA) o deleciones en ciertos cromosomas (deleciones en el cromosoma 18q cerca del gen DCC) pueden ser factores pronósticos independientes. El gen ki-ras regula, en condiciones normales, los factores de crecimiento celular. Alteraciones en este gen que conllevan una disregulación de su función se han detectado en aproximadamente el 50% de los CCR. Algunos autores han asociado esta mutación a un peor pronóstico y a una peor respuesta al tratamiento117. El gen p53 codifica una fosfoproteína nuclear que se comporta como reguladora de la transcripción. Deleciones a este nivel se han localizado en aproximadamente el 80% de los CCR. La mayoría de los estudios lo han asociado a un peor pronóstico y peor respuesta a tratamiento118-120. Los alelos del cromosoma 18q juegan un papel importante en la tumorogénesis gastrointestinal. Algunos autores han demostrado que los pacientes con CCR en estadio II que conservan los alelos del cromosoma 18q pueden tener una supervivencia media similar a los pacientes en estadio I, pero que aquellos pacientes en estadio II con pérdida de los alelos del cromosoma 18q pueden tener una supervivencia media a los 5 años similar a la de aquellos pacientes con CCR en estadio III121. Martínez-López et al. observaron que en 114 casos de CCR esporádico, aquéllos que presentaban deleciones del cromosoma 18q mostraban una supervivencia significativamente menor (42 versus 73%) a los 5 años de seguimiento122. Sin embargo, otros autores han demostrado que este parámetro no se comporta como un predictor independiente de pronóstico123, por lo que habrá que esperar nuevos resultados para poder extraer conclusiones fiables.
3. ¿Qué otros métodos existen para aumentar la sensibilidad y/o especificidad del CEA?
Existen tres estudios aleatorizados y controlados que han evaluado la efectividad del seguimiento intensivo del CCR con baterías de pruebas que incluyeran el CEA, si bien su propósito no era evaluar la utilidad de este marcador en dicho seguimiento. Los tres estudios encontraron que el seguimiento intensivo conllevó mayor incidencia de cirugía sin encontrarse finalmente beneficio alguno124.
Aproximadamente, tres cuartas partes de los pacientes con recurrencias de CCR presentan elevaciones en las concentraciones de CEA antes de que comiencen a desarrollar sintomatología125. Aunque es una práctica habitual en la medicina actual de terminar seriadamente el CEA en el seguimiento de los pacientes diagnosticados de CCR, la evidencia existente al respecto es contradictoria. A ello contribuye el que los distintos estudios sobre CEA publicados se hayan realizado con distintas técnicas de laboratorio, que las condiciones de preparación de las muestras sean distintas. Todo ello hace que sea difícil neutralizar diferencias de hasta el 50% entre los valores del CEA debidos exclusivamente a la aleatorización126,127.
--Evidencias a favor del CEA seriado en el seguimiento del CCR:
Bruinvels et al., en un metaanálisis de 3.283 pacientes incluidos en 7 estudios no aleatorizados compararon el seguimiento intensivo tras resección colorrectal con el seguimiento no intensivo o con el no-seguimiento. El seguimiento intensivo incluía historia clínica, exploración física y pruebas séricas, radiológicas y endoscópicas. Se detectaron más recurrencias asintomáticas en los pacientes con seguimiento intensivo (45 versus 8%), las cuales eran más frecuentemente resecables (35 versus 21%) y presentaron una supervivencia media a los 5 años 9% superior a los grupos control. Sin embargo, la ventaja en la supervivencia media fue significativa sólo en el grupo de pacientes con determinaciones de CEA en el seguimiento128.
Pietra et al. demostraron una diferencia significativa en términos de supervivencia a favor de los pacientes bajo seguimiento intensivo, siendo el CEA la prueba más importante para diagnosticar recidivas129.
Carriquriy et al. encontraron una sensibilidad del CEA del 77% y una especificidad del 98% en el diagnóstico de recurrencias de CCR, la mayoría de ellas (63%) asintomáticos, alcanzando sensibilidades del 92% cuando existían metástasis a distancia y casi del 100% cuando existían metástasis hepáticas, aunque descendió al 62% en el diagnóstico de recurrencias locales, recomendando determinaciones de CEA cada 3 meses durante los primeros 2 años, cada 4 meses durante los dos siguientes, y anualmente a partir de entonces y considerando anormal dos elevaciones consecutivas de CEA130.
-- Evidencias en contra del CEA seriado en el seguimiento del CCR:
Existen varios trabajos que evalúan la efectividad del CEA en el seguimiento del CCR.
Por una parte, Moertel et al. estudiaron en 1993 un total de 1.217 pacientes en el momento de recibir terapia adyuvante según se les determinase o no la concentración de CEA en plasma. De los 1.017 a quienes determinaron el CEA, 345 mostraron valores superiores a la normalidad. No encontraron diferencia alguna en la segunda cirugía entre el grupo con CEA elevado, CEA normal y CEA no determinado en cuanto a porcentaje de pacientes libres de enfermedad al año de seguimiento (2,9 vs. 1,9 vs. 2,0, respectivamente). Encontraron que el CEA presentaba tan sólo una sensibilidad del 59% para detectar recurrencias, con un 16% de falsos positivos. No hallaron diferencia alguna entre los distintos grupos en cuanto a mortalidad y supervivencia libre de enfermedad al año de seguimiento, cuestionado, a la luz de dichos resultados, que fuese eficiente la monitorización de CEA en el seguimiento de estos pacientes. El coste estimado por paciente curado en los EE.UU. asciende a aproximadamente 500.000 dólares131.
Por otra parte, Ohlsson et al. estudiaron prospectivamente, durante una media de 5,5 a 8,8 años, un grupo de pacientes sometido a cirugía curativa por CCR. Fueron aleatorizados a un grupo control (vistos en consulta sólo si presentaban síntomas) o a un grupo de seguimiento intensivo (exploración física, sangre oculta en heces, endoscopia, escáner, radiografía de tórax, pruebas hepáticas y determinación seriada de CEA). Ambos grupos presentaron recurrencias en un 30% de los casos. El mismo número de pacientes en ambos grupos fue sometido a cirugía curativa. No encontraron diferencias en la supervivencia a los 5 años entre los dos grupos132. No obstante, como señalan algunos autores, este trabajo incluyó un número escaso de sujetos, sin la potencia necesaria para aceptar con suficiente confianza que no se hayan pasado diferencias significativas133.
Finalmente, Northover et al. aleatorizaron 1.447 pacientes sometidos a cirugía potencialmente curativa a un grupo control y un grupo de intervención. Determinaron CEA periódicamente a todos los pacientes, condicionando, en el grupo de intervención, la elevación del CEA una ampliación del estudio, incluyendo laparotomía exploradora en los casos necesarios. No encontraron diferencias significativas en términos de supervivencia entre los dos grupos, interpretando que el CEA es un pobre predictor de recurrencia local y, a veces, también en recidivas sistémicas. Los resultados de este estudio, citado por numerosos autores, no han sido publicados en ningún medio científico, al menos hasta donde llega nuestro conocimiento. No obstante, parece ser que no era un estudio diseñado para estudiar el CEA en el CCR, aparte de que los propios autores admiten que consideraron como anormal cualquier valor elevado de CEA, independientemente de que fuese una elevación transitoria, lo cual desembocó en un número elevado de falsos positivos133.
Otra razón argumentada en contra de la monitorización del CEA en el seguimiento de pacientes con CCR es que se sabe que cerca del 30% de las recurrencias del CCR no conllevan incrementos del CEA134.
Los estudios de coste efectividad existentes tampoco parecen apoyar la monitorización del CEA en estos pacientes. Kievit et al. analizaron mediante análisis de decisión el beneficio de la monitorización del CEA en la esperanza de vida en pacientes con CCR. El beneficio en esperanza de vida ajustada por la calidad varió entre un decremento medio de 5 días y un incremento medio de 7 días. El coste asociado a cada año de vida ajustado por la calidad varió entre 22.963 y 4.888.208 dólares135.
Podemos concluir que el papel de la monitorización del CEA en el seguimiento del CCR sigue siendo controvertido. Hoy por hoy, no existe evidencia suficiente para recomendar la monitorización seriada de CEA en los pacientes diagnosticados de CCR. No obstante, la ASCO recomienda que se determine el CEA cada 2 ó 3 meses durante al menos los dos primeros años tras el diagnóstico de CCR en estadio II o III134. Una elevación aislada debe confirmarse analíticamente. Una vez confirmada, una ampliación del estudio está indicada (escáner abdominal y pélvico, radiografía de tórax y, en ocasiones, colonoscopia). Esta indicación sólo es válida en pacientes susceptibles de recibir cirugía que suponga un claro beneficio potencial136.
Actualmente, está abierta una nueva y prometedora vía de investigación sobre el CEA: los anticuerpos policlonales y monoclonales anti-CEA. Parece que han demostrado ser capaces de detectar tumores que incluso pasan desapercibidos en las pruebas de imagen137.
4. ¿Qué recomendaciones establecen los Grupos de Trabajo sobre los MT en el manejo y seguimiento del CCR?
Las recomendaciones de los grupos de trabajo sobre el empleo del los MT en el manejo y seguimiento del CCR se establecen en la tabla VII.138-140 .

De todos los MT valorados por los Grupos de Trabajo en el seguimiento del CCR (CA 19,9, LASA, índice de DNA, porcentaje de DNA en fase S, mutación o expresión de p53, el oncogen ras y CEA), sólo la monitorización de este último se recomienda, reservándolo para pacientes diagnosticados de CCR resecables en los que esta cirugía suponga un beneficio clínico potencial. Se recomienda cada 2-3 meses en los CCR estadio II o III durante al menos dos años tras el diagnóstico, pero con una fuerza de recomendacion C.
Cáncer de mama
Existen numerosos MT empleados en el seguimiento del CM ( II). Entre ellos destacan las citokeratinas, caseína, lactoalbúmina, SP-1, transferrina-lactoferrina, MEMA, antígeno T, TPA y TPS, actina y miosina, GCDFP-15, PEM, S-100, PSA u otros marcadores séricos (CA 15,3, CASA, MSA, CA 549, SP2, CA27,29), pero ninguno de ellos ha demostrado reunir las condiciones idóneas para convertirse en el marcador diagnóstico de elección. No existe evidencia que demuestre que las elevaciones en ellos precedan a la aparición del tumor con el suficiente tiempo de antelación para intervenir y alterar favorablemente el pronóstico141. Por ello, es preciso esperar a los resultados de nuevos estudios antes de extraer conclusiones.
En el seguimiento del CM, la ASCO recomienda realizar historia clínica y exploración física cada 3-6 meses durante los dos años siguientes al tratamiento, cada 6-12 meses durante los dos años después y anualmente a partir de entonces, y una mamografía basal a los 6 meses y al año de la operación y después anualmente142. Sin embargo, no recomienda la realización sistemática de pruebas analíticas, radiografía de tórax, escáner óseo ni determinación seriada de marcadores tumorales.
No existe evidencia científica de que ningún marcador tumoral aumente la supervivencia de los CM detectados, la calidad de vida o el balance coste-efectividad. Por ello, se necesitan más estudios antes de poder realizar otro tipo de recomendaciones.
1. ¿Existe algún marcador tumoral que pueda emplearse como factor pronóstico en el CM?
En el caso del CM, no existe en la actualidad ningún marcador que reúna características de utilidad como un factor pronóstico, pero existen algunos que se presentan como posibles marcadores pronósticos de futuro:
--Receptores de estrógenos (RE) y progesterona (RP): la determinación de los RE y RP se ha convertido en rutinaria en la práctica clínica diaria con los pacientes con CM, debido a que parecen tener cierta importancia a la hora de predecir la respuesta al tratamiento hormonal. Aún queda por resolver cuál es la mejor forma de dar los resultados (considerar positivo a cualquier tumor que tiña de receptor más del 5% de sus células, tener en cuenta la intensidad de la tinción y el número de células teñidas, emplear técnicas de imagen, etc.). Independientemente de que un resultado positivo para RE pueda tener significado pronóstico en algunos tipos de tumores (algunos carcinomas ductales in situ), actualmente no existe evidencia científica que apoye la determinación rutinaria de receptores de hormonas con este fin en los pacientes con CM143.
--Contenido de DNA: independientemente de que la aneuploidía celular se relacione habitualmente con algunos factores de peor pronóstico (baja diferenciación celular, tamaño del tumor, existencia de ganglios axilares, estadio, ausencia de RE, etc.), no existe suficiente evidencia que sugiera que el contenido de DNA constituye un importante factor pronóstico independiente en pacientes con CM144-146.
--Velocidad de crecimiento: existen numerosos estudios que han demostrado que la determinación de la velocidad de crecimiento o proliferación tumoral es un factor pronóstico independiente en los pacientes con CM144-147.
--Oncogén HER-2/NEU: aproximadamente el 15-30% de los CM presentan expresión incrementada del oncogén Her 2/neu (C-erbB-2). Existen estudios tanto a favor como en contra de que este oncogén constituya un factor pronóstico independiente en estos pacientes. Existe evidencia de que este oncogén identifica un subgrupo de pacientes que mostrarán buena respuesta al tratamiento quimioterápico, por lo que es un marcador a tener en cuenta hoy en día y con grandes esperanzas depositadas en él para el futuro148.
--p53: aproximadamente el 20-50% de los pacientes con CM presentan mutaciones del gen supresor de tumores p53 o acúmulo de esta proteína. El porcentaje es aún mayor entre pacientes con cáncer hereditario o familiar. Numerosos estudios prospectivos han puesto de manifiesto que esta proteína es un factor pronóstico independiente de supervivencia total y de intervalo libre de enfermedad tanto en CM con ganglios positivos como negativos149-150. No obstante, existen otros estudios en los que no se ha encontrado esta asociación145, 151. Por lo tanto, en ausencia de un consenso sobre el uso del gen p53 como factor pronóstico, es necesario esperar nuevos resultados para poder realizar conclusiones en este sentido.
--Catepsina D: existen estudios que han relacionado la existencia de esta proteasa con un peor pronóstico en los pacientes con CM145, aunque las distintas técnicas de detección de la misma dificultan la homogeneización de criterios. Por lo tanto, es necesario esperar nuevos resultados que apoyen o rechacen el uso de esta proteasa como factor pronóstico independiente en pacientes con CM.
--Angiogénesis tumoral: no existe consenso sobre su aplicación presente como marcador de pronóstico, si bien hay grandes esperanzas puestas en sus marcadores para un próximo futuro145, 152,153.
Podemos concluir que los únicos factores pronósticos de la evolución del CM aceptados universalmente son la afectación ganglionar, el tamaño, la histología y el estadiaje del tumor. No existe evidencia suficiente para apoyar unilateralmente ningún otro marcador como factor pronóstico independiente de la evolución del CM154.
Aplicación de los resultados a la práctica clínica en Atención Primaria. Conclusiones
1. No se recomienda el cribado sistemático para cáncer de próstata mediante PSA. De llevarse a cabo, la mayor parte de autores y grupos de trabajo recomiendan a día de hoy la utilización de PSA y tacto rectal.
2. No se recomienda el screening de cáncer de ovario mediante MT, ecografía transvaginal y/o exploración pélvica en mujeres asintomáticas, y no existe evidencia suficiente para hacer recomendaciones a favor o en contra del cribado de cáncer de ovario en mujeres pertenecientes a grupos de riesgo.
3. No se recomienda el screening de cáncer colorrectal mediante MT.
4. No se recomienda el screening de cáncer de mama mediante MT.
5. Se recomienda la monitarización de PSA y tacto rectal en la detección de recurrencias de cáncer de próstata.
6. Se recomienda la determinación de CA 125 en el seguimiento de la respuesta al tratamiento quirúrgico y quimioterápico y aparición de recidivas en el cáncer de ovario.
7. No se recomienda el uso de MT en el manejo y seguimiento del cáncer colorrectal, excepto el CEA en el caso de CCR resecables en los que la cirugía suponga un beneficio clínico potencial, pero con fuerza de recomendación C.
8. No existe evidencia científica de que la determinación sistemática de MT aumente la supervivencia de los CM detectados, la calidad de vida o el balance coste-efectividad.
CORRESPONDENCIA:
Gustavo de Teresa Romero
C.S. Parquesol
C/ Ciudad de la Habana, s/n
47014 Valladolid
e-mail: gustavodt@yahoo.com
Bibliografía
1. Ministerio de Sanidad y Consumo. Indicadores de Salud. Tercera evaluación en España del programa regional europeo. Salud para todos. Madrid: Ministerio de Sanidad y Consumo, 1995. [ Links ]
2. Valle MO, López ML, Arcos PI. Cueto A. Análisis de los años potenciales de vida perdidos por cáncer en Asturias y España. Rev Hig Publica 1993; 67: 129-44. [ Links ]
3. López-Abente G. Atlas de Mortalidad por Cáncer y otras causas en España. 1978-92. 1996. Instituto Nacional de Estadística. Defunciones según la causa de muerte. 1993. Tomo 1. Resultados básicos. Madrid: INE, 1996. [ Links ]
4. Cortés H, Díaz Rubio E, García Conde J, Germá Lluch JR, Guillén Porta V, López López J. Oncología Médica. Madrid: Aula Médica SA, 1999. [ Links ]
5. Gálvez Ibáñez. Marcadores tumorales. Utilización por el médico de familia. FMC 2000; 7 (Supl 5): 21-6. [ Links ]
6. Molina R, Filella X, Ballesta AM. Marcadores tumorales. Utilidad clínica en el cáncer de mama y en neoplasias epiteliales ováricas. FMC 2000; 7 (Supl 5): 30-5. [ Links ]
7. Wilson JMG, Jungner F. Principles and practice of screening for disease. Public Health Papers nº 34. Geneva: World Health Organization, 1968. [ Links ]
8. Sackett, Richardson, Rosenberg, Haynes. Evidence-Based Medicine: How to practice and teach EBM. London: Curchill Livingstone, 1997. [ Links ]
9. Greenhalgh T. How to read a paper: Papers that report diagnostic or screening tests. BMJ 1997; 315: 540-3. [ Links ]
10. Canadian Task Force on the Periodic Health Examination, Anonymous. The periodic health examination. Canadian Task Force on the Periodic Health Examination. Can Med Assoc J 1979; 121: 1193-254. [ Links ]
11. Programa de Actividades Preventivas y Promoción de la Salud (PAPPS). Sociedad Española de Medicina de Familia y Comunitaria (semFYC). Barcelona, 1999. [ Links ]
12. Gohagan JK, Prorok PC, Kramer BS, Cornett JE. Prostate cancer screening in the prostate, lung, colorectal and ovarian cancer screening trial of the National Cancer Institute. Journal of Urology 1994; 152: 1905-9. [ Links ]
13. Shroder FH, Bangma CH. The European Randomised study of screening for prostate cancer (ERSPC). Br J Urol 1997; 79: 68-71. [ Links ]
14. Labrie F, Candas B, Dupont A, Cusan L, Gomez J-L, Suburu RE, et al. Screening decreases prostate cancer death: first analysis of the 1988 Quebec prospective randomised controlled trial. Prostate 1999; 39: 83-91. [ Links ]
15. Alexander FE, Prescott RJ. Reply to Labrie et al. [Letter]. Prostate 1999; 40: 135-6. [ Links ]
16. Boer R, Schroder FH. Quebec randomised controlled trial on prostate cancer screening shows no evidence for mortality reduction. [Letter]. Prostate 1999; 40: 130-1. [ Links ]
17. Hara M, Koyanagi Y, Inoue T, Fukuyama T. Some physico-chemical characteristics of gamma seminoprotein, an antigen component specific for human seminal plasma. Nippon Hoigaku Zasshi 1971; 25: 322-4. [ Links ]
18. Wang MC, Valenzuela LA, Murphy GP, Chu TM. Purification of a human prostate specific antigen. Investigative Urology 1979; 17: 159-63. [ Links ]
19. Diamandis EP, Yu H. Editorial: new biological functions of prostate-specific antigen? J Clin Endocrinol and Metab 1995; 80(5): 1515-7. [ Links ]
20. Takayama TK, Vessela RL, Brawer MK, True LD, Notebom J, Lange PH. Urinary PSA levels after radical prostatectomy. J Urol 1994; 151: 82-7. [ Links ]
21. Ellis WJ, Vessela RL, Notebom JL, Lange PH, Wolfert RL, Rittenhouse HG. Early detection of recurrent prostate cancer with an ultrasensitive chemiluminescent prostate specific antigen assay. Urology 1997; 50: 573-9. [ Links ]
22. Herschmann JD, Smith DS, Catalona WJ. Effect of ejaculation on serum prostate specific antigen concentrations. Urology 1997; 50: 239-43. [ Links ]
23. American Urological Association. Prostate-specific antigen (PSA) Best Practice Policy. Oncology 2000; 14(2): 267-86. [ Links ]
24. Catalona WJ, Richie JP, Ahmann FR, Hudson MA, Scardino PT, Flanigan RC, et al. Comparison of digital rectal examination and serum prostate specific antigen in the early detection of prostate cancer: results of a multicentre clinical trial of 6630 men. J Urol 1994; 151: 1283-90. [ Links ]
25. Optenberg SA, Thompson IM. Economics of screening for carcinoma of the prostate. Urol Clin North Am 1990; 17: 719-37. [ Links ]
26. Brawer MK. Prostate-specific antigen: Current status. CA Cancer J Clin 1999; 49(5): 264-81. [ Links ]
27. Gann PH, Henneknes CH, Stampfer MJ. A prospective evaluation of plasma prostate specific antigen for detection of prostate cancer. JAMA 1995; 273: 289-94. [ Links ]
28. Partin AW, Carter HB, Chan DW, Epstein JI, Oesterling JE, Rock RC, et al. Prostate-specific antigen in the staging of localised prostate cancer.: influence of tumour differentiation, tumour volume and benign hyperplasia. J Urol 1990; 143: 747-52. [ Links ]
29. Lange PH, Ercole CJ, Lightner DJ, Fraley EE, Vessella R. The value of serum prostate specific antigen determinations before and after radical prostatectomy. J Urol 1989; 141: 873-9. [ Links ]
30. Catalona WJ, Smith DS, Ornstein DK. Prostate cancer detection in men with serum PSA concentrations of 2,6 to 4,0 ng/ml and benign prostate examination. Enhancement of specificity with free PSA measurements. JAMA 1997; 277: 1452-8. [ Links ]
31. Stenman UH, Leinonen J, Ranniko S, Tuhkanen K, Alfthan O. A complex between prostate specific antigen and alpha-1-antichymotrypsin is the major form of prostate specific antigen in serum of patients with prostatic cancer. Assay of complex improves in clinical sensitivity for cancer. Cancer Research 1991; 51: 221-6. [ Links ]
32. Polascik TJ, Oesterling JE, Partin AW. PSA: A decade of discovery -What we have learned and where we are going-. J Urol 1999; 162(2): 293-306. [ Links ]
33. Benson MC, Whang IS, Pantuck A, Ring K, Kaplan SA, Olsson CA, et al. Prostate specific antigen density: a ways of distinguishing benign prostatic hypertrophy and prostate cancer. J Urol 1992; 147: 847-8. [ Links ]
34. Nishiya M, Miller GJ, Looknner DH. Prostate specific antigen density in patients with histologically proven prostate carcinoma. Cancer 1994; 75(11): 3002. [ Links ]
35. Semjonow A, Hamm M, Rathert P, Hertle L. Prostate-specific antigen corrected for prostate volume improves differentiation of benign prostatic hyperplasia and organ-confined prostatic cancer. Br J Urol 1994; 73: 538-43. [ Links ]
36. Bangma CH, Grobbee DE, Schroder FH. Volume adjustment of intermediate prostate specific antigen values in a screening population. Eur J Cancer 1995; 31A(1): 12-4. [ Links ]
37. Babaian RJ, Kojima M, Ramirez EI. Comparative analysis of prostate specific antigen and its indexes in the detection of prostate cancer. J Urol 1996; 156(2): 432-7. [ Links ]
38. Keetch DW, McMurtry JM, Smith DS, Andriole GL, Catalona WJ. Prostate specific antigen density versus prostate specific antigen slope as predictors of prostate cancer in men with initially negative prostate biopsies. J Urol 1996; 156: 428-31. [ Links ]
39. Carter HB, Pearson JB, Metter EJ, Brant LJ, Chan DW, Andres R, et al. Longitudinal evaluation of prostate specific antigen levels in men with and without prostate disease. JAMA 1992; 267: 2215-20. [ Links ]
40. Woolf S. Screening for prostate cancer with prostate specific antigen: an examination of evidence. N Engl J Med 1995; 333: 1401-5. [ Links ]
41. Prestigiacomo AF, Lilja H, Petterson K, Wolfert RL, Stamey TA. A comparison of the free fraction of serum prostate specific antigen in men with benign and cancerous prostates: the best case scenarios. J Urol 1996; 156: 350-6. [ Links ]
42. Brawer MK, Aramburu EAG, Chen GL, Preston SL, Ellis WJ. The inability of prostate specific antigen index to en-hance the predictive value of prostate specific antigen in the diagnosis of prostate carcinoma. J Urol 1993; 150: 369-73. [ Links ]
43. Carter HB, Pearson ID, Waclawiw Z, Metter EJ, Chan DW, Guess HA, et al. Prostate-specific antigen variability in men without prostate cancer: effect of sampling interval on prostate-specific antigen velocity. Urology 1995; 45(4): 591-6. [ Links ]
44. Kadmon D, Weinberg A, Williams RH, Pavlik VN, Cooper P, Migliore PJ. Pitfalls in interpreting prostate specific antigen velocity. J Urol 1996; 155: 1655-7. [ Links ]
45. Oesterling JE, Jacobsen SJ, Chute CG, Guess HA, Girman CJ, Panser LA, et al. Serum prostate specific antigen in a community based population of healthy men: establishment of age-specific reference ranges. JAMA 1993; 270: 860-4. [ Links ]
46. Oesterling JE, Jacobsen SJ, Klee CG, Petterson K, Piironen T, Abrahamsson PA, et al. Free, complex and total prostate specific antigen: the establishment of appropriate reference ranges for their concentrations and ratios. J Urol 1995; 154: 1090-5. [ Links ]
47. Partin AW, Criley SR, Subong EN, Zincke H, Walsh PC, Oesterling JE. Standard versus age-specific reference ranges among men with clinically localised prostate cancer: a pa-thological analysis. J Urol 1996; 155: 1336-9. [ Links ]
48. Catalona WJ, Smith DS, Wolfert R, Wang TJ, Rittenhouse H, Ratliff TL, et al. Evaluation of percentage of free serum prostate specific antigen to improve specificity of prostate cancer screening. JAMA 1995; 274:1214-20. [ Links ]
49. Petteway JC, Brawer M. Age specific versus 4,0 ng/ml as PSA cutoff in the screening population: impact on cancer detection. J Urol 1995; 153(suppl): 465A-472A. [ Links ]
50. Epstein JL, Walsh PC, Carmichael M, Brendler CB. Pathologic and clinical findings to predict tumour extent of non-palpable (stage T1c) prostate cancer. JAMA 1994; 271(5): 368-74. [ Links ]
51. Dugan JA, Bostwick DG, Myers RP. The definition and preoperative prediction of clinically insignificant prostate cancer. JAMA 1996; 275(44): 288-94. [ Links ]
52. Mettlin C, Murphy GP, Lee F, Littrup PJ, Chesley A, Babaian R. Characteristics of prostate cancer detected in the American Cancer Society-National Prostate Cancer Detection Project. J Urol 1994; 152: 1737-40. [ Links ]
53. Partin AW, Kattan MW, Subong ENP. Combination of prostate specific antigen, clinical stage, and Gleason score to predict pathological stage of localised prostate cancer: a multi-institutional update. JAMA 1997; 277(18): 1445-51. [ Links ]
54. American Cancer Society. ACS prostate screening guide-line 1997. Update of the 1993 recommendations. Cancer J Clin 1997; 47(5): 261-4. [ Links ]
55. American College of Physicians-American Society of Internal Medicine (ACP-ASIM). Early detection of prostate cancer. Ann Intern Med 1997; 126: 394-406. [ Links ]
56. Canadian Urological Association. Guidelines for early detection of prostate cancer. Can J Oncol 1994; 4(S1): 82-5. [ Links ]
57. National Cancer Institute Physician Statement: Screening for prostate cancer. (Updated may 2000). National Cancer Institute report (www.nci.nih.gov). [ Links ]
58. US Preventive Services Task Force (2nd ed). Guide to clinical preventive services. Baltimore: US Preventive Services Task Force. Williams and Wilkins, 1996. [ Links ]
59. Canadian Task Force on the Periodic Health Examination. The Canadian Guide to Clinical Preventive Health Care. Ottawa: Canada Communication Group, 1994. [ Links ]
60. Green CJ, Hadorn D, Basset K, Kazanzian A. Prostate specific antigen in the early detection of prostate cancer. BC Office of Health Technology Assessment discussion paper series. Canadian Cancer Society. Vancouver: BCOHTA 1993; 93: 1-66. [ Links ]
61. NHS Centre for Reviews and Dissemination. Screening for prostate cancer. Effectiveness Matters 1997; 2: (2). (www.york.ac.uk/inst/crd/em22a.htm) [ Links ]
62. The Royal College of Radiologists' Clinical Oncology Information Network. British Association of Urological Sur-geons. Guidelines for the management of prostate cancer. BJU International 1999; 84: 987-1014. [ Links ]
63. Bast RC, Klug TL, St John E, Jenison E, Lazarus H, Berkowitz RS, et al. Radioimmunoassay using a monoclonal antibody to monitor the course of epithelial ovarian cancer. N Engl J Med 1983; 309: 883-7. [ Links ]
64. Haga Y, Sakamoto K, Egami H. Evaluation of serum CA 125 values in healthy individuals and pregnant women. Am J Med Sci 1986; 292: 25-9. [ Links ]
65. Jacobs I, Bast RC. The CA 125 tumour-associated antigen: a review of the literature. Human Reproduction 1989; 4: 1-12. [ Links ]
66. Ovarian Cancer: Screening, Treatment and Follow-up. NIH Consensus Development Conference Statement. 1994. April 5-7;12(3):30. Reviewed on 1998. [ Links ]
67. Bell R, Petticrew M, Sheldon T. The performance of screening tests for ovarian cancer: results of a systematic review. Br J Obstet Gynaecol 1998; 105: 1136-47. [ Links ]
68. Jacobs IJ, Skates SJ, McDonald N, Menon U, Rosenthal AN, Davies AP, et al. Screening for ovarian cancer: a pilot randomised controlled trial. Lancet 1999; 353: 1207-10. [ Links ]
69. Jacobs I, Skates SJ, Pryse-Davis J. Risk of diagnosis of ovarian cancer after raised serum CA 125 concentration: a prospective cohort study. BMJ 1993; 306: 1030-40. [ Links ]
70. MacDonald ND, Jacobs IJ. Is there a place for screening in ovarian cancer? Eur J Obstet Gynecol Reprod Biol 1999; 82: 155-7. [ Links ]
71. Jacobs IJ, Skates S, Davies AP, Woolas RP, Yeyerajah A, Weidermann P, et al. Risk of diagnosis of ovarian cancer after raised serum CA 125 concentration: a prospective cohort study. BMJ 1996; 313: 1355-8. [ Links ]
72. Helzllsouer KJ, Bush TL, Alberg AJ, Bass KM, Zacer H, Comstock GW. Prospective study of serum CA 125 levels as markers of ovarian cancer. JAMA 1993; 269: 1123-6. [ Links ]
73. NHS Centre for Reviews and Dissemination. Management of Gynaecological cancers. Effective Health Care 1999; 5(3): 1-12. [ Links ]
74. Jacobs I, Davies AP, Bridges J, Stabile I, Fay T, Lower A. et al. Prevalence screening for ovarian cancer in postmenopausal women by CA 125 measurement and ultrasonography. BMJ 1993; 306: 1030-4. [ Links ]
75. Jacobs I, Stabile I, Bridges J, Kemsley P, Reynolds C, Grudzinskas J, et al. Multimodal approach to screening for ovarian cancer. Lancet 1988; 1: 268-71. [ Links ]
76. Karlan BY, Raffel LJ, Crvenkovic G, Smrt C, Chen MD, Lopez E, et al. A multidisciplinary approach to the early detection of ovarian carcinoma: rationale, protocol design, and early results. Am J Obstet Gynecol 1993; 169: 494-501. [ Links ]
77. Nguyen HN, Averette HE, Janicek M. Ovarian carcinoma. A review of the significance of familial risk factors and the role of prophylactic oophorectomy in cancer prevention. Cancer 1994; 74: 545-55. [ Links ]
78. Skates SJ, Singer DE. Quantifying the potential benefit of CA 125 screening for ovarian cancer. J Clin Epidemiol 1991; 44: 365-80. [ Links ]
79. Schapira MM, Matchar DB, Young MJ. The effectiveness of ovarian cancer screening: a decision-analysis model. Ann Intern Med 1993; 118: 838-43. [ Links ]
80. Piver MS, Goldberg JM, Tsukada Y. Characteristics of familial ovarian cancer: a report of the first 1000 families in the Gilda Radner Familial Ovarian Cancer Registry. European Journal of Gynaecologic Oncology 1996; 17(3): 169-76. [ Links ]
81. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harschman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA 1. Science 1994; 266: 66-71. [ Links ]
82. Easton DF, Bishop DT, Ford D, Crockford GP. Genetic linkage analysis in familial breast and ovarian cancer: results form 214 families. American Journal of Human Genetics 1993; 52: 678-701. [ Links ]
83. Easton DF, Ford D, Bishop DT. Breast and ovarian cancer incidence in BRCA 1-mutation carriers. Breast Cancer Linkage Consortium. American Journal of Human Genetics 1995; 56: 265-71. [ Links ]
84. Struewing JP, Hartge P, Wacholder S, Baker SM, Berlin M, McAdams M, et al. The risk of cancer associated with specific mutations of BRCA 1 and BRCA 2 among Ashkenazi Jews. New Eng J Med 1997; 336: 1401-8. [ Links ]
85. Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, et al. Localization of a breast cancer susceptibility gene, BRCA 2, to chromosome 13q12-13. Science 1994; 265: 2088-90. [ Links ]
86. NHS Centre for Reviews and Dissemination. Screening for ovarian cancer: a systematic review (CRD 13). (www.york.ac.uk/inst/crd/report13). [ Links ]
87. American College of Obstetrics and Gynecology Committee on Practice Bulletins. Routine cancer screening. ACOG Committee Opinion 1997; 185: 225-9. [ Links ]
88. American College of Obstetrics and Gynecology Committee on Educational Bulletins. Ovarian cancer. Educ Bull 1998; 250: 667-75. [ Links ]
89. Burke W, Daly M, Garber J, Botkin J, Kahn MJ, Lynch P, et al. Recommendations for follow-up care on individuals with an inherited predisposition to cancer. II. BRCA 1 and BRCA 2. JAMA 1997; 277: 997-1003. [ Links ]
90. Olopade OI, Lynch PM. Genetic testing, guidelines, ethics -when to test and what to do with the results. ASCO 1997; 16: 58-62. [ Links ]
91. Oesterling JE, Martin SK, Bergstralh EJ. The use of PSA in staging patients with newly diagnosed prostate cancer. JAMA 1993; 269: 57-60. [ Links ]
92. Vessela RL, Lange PH. Issues in the measurement of PSA inmunoassays. Urol Clin North Am 1993; 20: 607-19. [ Links ]
93. Stein A, de Kernion JB, Smith RB, Dorey F, Patel H. Prostate specific antigen levels after radical prostatectomy in patients with organ-confined and locally extensive prostate cancer. J Urol 1992; 147: 942-6. [ Links ]
94. Yu H, Diamandis EP, Wong PY, Nann R, Trachtenberg J. Detection of prostate cancer relapse with prostate specific antigen monitoring at levels of 0,0001 to 0,1 mcg/l. J Urol 1997; 157-913-8. [ Links ]
95. Catalona WJ, Smith DS. 5-year tumour recurrence rates after anatomical radical retropubic prostatectomy for prostate cancer. J Urol 1994; 152: 1837-44. [ Links ]
96. Trapasso JG, deKernion JB, Smith RB, Dorey F. The incidence and significance of detectable levels of serum pros-tate specific antigen after radical prostatectomy. J Urol 1994; 152: 1821-5. [ Links ]
97. Fichtner I. The management of prostate cancer in patients with a rising prostate-specific antigen level. BJU International 2000; 86: 181-90. [ Links ]
98. Canadian Coordinating Office for Health Technology Assessment. Predictive genetic testing for breast and prostate cancer. The Cochrane Collaboration. CRD database number: HTA-998425. [ Links ]
99. Zietman AL, Coen JJ, Shipley WU. Radical radiation therapy in the management of prostatic adenocarcinoma: The initial prostate specific antigen value as a predictor of treatment outcome. Cancer 1994; 151: 640-5. [ Links ]
100. Critz FA, Levinson AK, Williams WH. Prostate-specific antigen nadir: The optimum level after irradiation for prostate cancer. J Clin Oncol 1996; 149: 2893-900. [ Links ]
101. American Society of Therapeutic Radiology and Oncology Consensus Panel. Consensus statement: Guidelines for PSA following radiation therapy. Int J Radiat Oncol Biol Phys 1997; 37: 1035-40. [ Links ]
102. Carducci MA, DeWeese TL, Nelson JB. Prostate-specific antigen and other markers of therapeutic response. Urol Clin of North America 1999; 2: 291-302. [ Links ]
103. Brausi M, Jones WG, Fossa SD. High dose epirubicin is effective in measurable metastatic prostate cancer. A phase II study of the EORTC genitourinary group. Eur J Cancer 1995; 31A: 1622-6. [ Links ]
104. Dawson MA, McLeod DG. The assessment of treatment outcomes in metastatic prostate cancer: changing endpoints. Eur J Cancer 1997; 30: 560-5. [ Links ]
105. Meyer T, Rustin GJ. Role of tumour markers in monitoring epithelial ovarian cancer. Br J Cancer 2000; 82: 1535-8. [ Links ]
106. Maggino T, Gadducci A. Serum markers as prognostic factors in epithelial ovarian cancer: an overview. Eur J Gynaecol Oncol 2000; 21: 64-9. [ Links ]
107. Oehler MK, Sutterlin M, Caffier H. CASA and Ca 125 in diagnosis and follow-up of advanced ovarian cancer. Anticancer Res 1999; 2513-8. [ Links ]
108. Lukacsko I, Hernadi Z, Sapy T, Borsos A. The prognostic value of CA-125 in epithelial ovarian cancer patients during and after chemotherapy. Acta Chir Hung 1997; 36: 213-4. [ Links ]
109. de Bruijn HW, van der Zee AG, Aalders JG. The value of cancer antigen 125 (CA 125) during treatment and follow-up of patients with ovarian cancer. Curr Opin Obstet Gynecol 1997; 9: 8-13. [ Links ]
110. Maggino T, Gadducci A. Serum markers as prognostic factors in epithelial ovarian cancer: an overview. Eur J Gynaecol Oncol 2000; 21: 64-9. [ Links ]
111. Oehler MK, Sutterlin M, Caffier H. CASA and Ca 125 in diagnosis and follow-up of advanced ovarian cancer. Anticancer Res 1999; 2513-8. [ Links ]
112. Gold P, Freedman SO. Demonstration of tumour specific antigens in human colonic carcinomata by inmunologic tolerance and absorption techniques. J Exp Med 1965; 121: 439-66. [ Links ]
113. Wanebo JH, Rao G, Pinsky CM, Hoffman RG, Sterns M, Schwartz MK, et al. Preoperative CEA level as a prognostic indicator in colorectal cancer. N Engl J Med 1978; 299: 448-51. [ Links ]
114. Fielding LP. Pettigrew: College of American Pathologists Conference XXVI on clinical relevance of prognostic markers in solid tumours: Report of the colorectal cancer working group. Arch Pathol Lab Med 1995; 119: 115-21. [ Links ]
115. Slentz K, Senagore A, Hibbert J, Mazier WP, Talbot TM. Can preoperative and postoperative CEA predict survival after colon cancer resection?. Am Surg 1994; 60: 528-32. [ Links ]
116. Pokorny RM, Hount LE, Glandiuk S. What's new with tumour markers for colorectal cancer?. Dig Surg 2000; 17: 209-215. [ Links ]
117. Ahnen DJ, Feigi P, Quan G. Prognostic significance of allellic loss at chromosome 18q21 for stage II colorectal cancer. Gastroenterology 1998; 114: 1188-95. [ Links ]
118. Hamelin R, Laurent-Puig P, Olshwang S, Jego N. Association of p53 mutations with short survival in colorectal cancer. Gastroenterology 1994; 106: 42-8. [ Links ]
119. Goh HS, Yao J, Smith DR. P53 point mutations and survival in colorectal cancer patients. Cancer Res 1995; 55: 5217-21. [ Links ]
120. Lowe SW, Bodis S, McClatchey A, Remington L. P53 status and the efficacy of cancer therapy in vivo. Science 1994; 266: 807-10. [ Links ]
121. Jen J, Kim H, Piantadosi S. Allelic loss of chromosome 18q and prognosis in colorectal cancer. N Engl J Med 1994; 331: 213-21. [ Links ]
122. Martínez-López E, Abad A, Monzo M. Allelic loss on chromosome 18q as a prognostic marker in stage II colorectal cancer. Gastroenterology 1998; 114: 1188-95. [ Links ]
123. Carethers JM, Hawn JK, Greenson JK. Prognostic significance of allelic loss at chromosome 11q21 for stage II colorectal cancer. Gastroenterology 1998; 114: 1188-5. [ Links ]
124. NHS Centre for Reviews and Dissemination, University of York. Effective Health Care. The management of colorectal cancer, 1997; 3(6): 1-11. [ Links ]
125. McArdle C. ABC of colorectal cancer. Effectiveness of follow-up. BMJ 2000; 321: 1332-5. [ Links ]
126. Touitou Y, Haus E. Biologic rhythms in clinical and laboratory medicine. Springer-Verlag. 2nd/ed. Heidelberg, 1994. [ Links ]
127. Watine J, Miédougé M. Pre-analytical Variation of Laboratory Variables. Clin Chem Lab Med 2000; 38: 263. [ Links ]
128. Bruinvels DJ, Stiggelbout AM, Kievit J, van Houlwelingen HC, Habbema JD, van der Velde CJ. Follow-up of patients with colorectal cancer. A meta-analysis. Ann Surg 1994; 219(2): 174-82. [ Links ]
129. Pietra N, Sarli S, Costi R, Ouchemi C, Grattarola M, Peracchia A. Role of follow-up in management of local recurrences of colorectal cancer. Dis Col Rect 1998; 41: 1127-33. [ Links ]
130. Carriquiry LA, Pineyro A. Should carcinoembryonic antigen be used in the management of patients with colorectal cancer Dis Col Rect 1999; 42: 921-9. [ Links ]
131. Moertel CG, Fleming TR, MacDonald JS, Haller DG, Laurie JA, Tanngen C. An evaluation of CEA test monitoring patients with resected colon cancer. JAMA 1993; 270: 943-7. [ Links ]
132. Ohlsson B, Breland U, Ekberg J, Graffner H, Tranberg KG. Follow-up after curative surgery for colorectal carcinoma. Dis Colon Rectum 1995; 38: 619-26. [ Links ]
133. Carriquiry LA. A new opportunity for CEA. BMJ 2000; 321: 1347-48. [ Links ]
134. Desch CF, Benson AB, Smith TJ. Recommended colorectal cancer surveillance guidelines by the American Society of Clinical Oncology. J Clin Oncology 1999; 17: 1312-8. [ Links ]
135. Kievit J, Vam de Velde CJ. Utility and cost of CEA monitoring in colon cancer follow-up evaluation: a Markov analysis. Cancer 1990; 65: 2580-7. [ Links ]
136. Berman JM, Cheung RJ, Weinberg DS. Surveillance after colorectal resection. Lancet 2000; 355: 395-401. [ Links ]
137. Patt YZ, Podoloff DA, Curley S, Kasi L, Smith R, Bhadkamkar V, et al. Technetium 99m-labeled IMMU-4, a monoclonal antibody against CEA, for imaging of occult recurrent colorectal cancer in patients with rising serum CEA levels. J Clin Oncol 1994; 12: 489-95. [ Links ]
138. Desch CE, Benson AB 3rd, Smith TJ, Flynn PJ, Krause C, Loprinzi CL, et al. Recommended colorectal cancer surveillance guidelines by the American Society of Clinical Oncology. J Clin Oncol 1999; 17(4): 1312-21. [ Links ]
139. Recommended colorectal cancer surveillance guidelines by the American Society of Clinical Oncology (ASCO). Up-date of the 1999 guidelines. National Guideline Clearinghouse on line. J Clin Oncol 2000; 15(18): 3586-8. [ Links ]
140. National Cancer Institute Information from PDQ for Health Professionals. National Cancer> Institute database. 1999. Update December 2000.
141. Hayes DF. Serum (cirulatin) tumour markers for breast cancer. Recent Results Cancer Res 1996; 140: 101-13. [ Links ]
142. Smith TJ, Davidson NE, Schapira DV. American Society of Clinical Oncology 1998 update recommended breast cancer surveillance guidelines. J Clin Oncol 1999; 17: 1080-2. [ Links ]
143. Bur M, Zimarowski MJ, Schmitt SJ. Strogen receptor inmunohistochemistry in carcinoma in situ of the breast. Cancer 1992; 69: 1174-81. [ Links ]
144. Merkwlo DE, Winchester DJ, Goldshmidt RA. DAN flow cytometry and pathologic grading as prognostic guides in axillary lymph node-negative breast cancer. Cancer 1994; 74: 381-7. [ Links ]
145. Mansour EG, Ravdin PM, Dressler L. Prognostic factors in early breast cancer. Cancer 1994; 74: 381-400. [ Links ]
146. Hedley DW, Clark GM, Cornelisse CJ. Consensus review of the clinical utility of DNA cytometry in carcinoma of the breast. Breast Cancer Res Treat 1993; 28: 55-9. [ Links ]
147. Silvestrini R, Daidone MG, Luisi A. Biologic and clinicopa-thologic factors as indicators of specific relapse types in node-negative breast cancer. J Clin Oncol 1995; 13: 697-704. [ Links ]
148. Muss HG, Thor AD, Berry DA. C-erb-2 expression and response to adjuvant therapy in women with node-positive early breast cancer. N Engl J Med 1994; 330: 1260-6. [ Links ]
149. Elledge RM., Fuqua S, Clark GM. The role and prognostic significance of p53 gene alterations in breast cancer. Breast Cancer Res Treat 1993; 27: 95-102. [ Links ]
150. Barnes DM, Dublin EA, Fisher CJ. Inmunohistochemical detection of p53 protein in mammary carcinoma. An important new indepent indicator of prognosis?. Hum Pathol 1993; 24: 469-76. [ Links ]
151. Rosen PP, Lesser ML, Arroyo CD. P53 in node-negative breast carcinoma: An inmunohistochemical study of epidemiologic risk factors, histologic features, and prognosis. J Clin Oncol 1995; 13: 821-30. [ Links ]
152.Gasparini G, Harris AL. Clinical importance of the determination of tumour angiogenesis in breast carcinoma. Much more than a new prognostic tool. J Clin Oncol 1995; 13: 765-82. [ Links ]
153. Horak ER, Leek R, Klenk N. Angiogenesis asessed by platelet/endothelial cell adhesion molecule antibodies as indicator of node metastases and survival in breast cancer. Lancet 1992; 340: 1120-4. [ Links ]
154. Eisso S, Shoman S. Tumour markers. London: Chapman & Hall, 1998. [ Links ]

