










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkMedicina Oral, Patología Oral y Cirugía Bucal (Internet)
versión On-line ISSN 1698-6946
Med. oral patol. oral cir.bucal (Internet) vol.11 no.1 ene./feb. 2006
ORAL MEDICINE AND PATHOLOGY
Adamantiades-Behçet disease: An enigmatic process with oral manifestations
Asier Eguia 1, Mariana Villarroel 2, Rafael Martínez-Conde 1, María Ángeles Echebarría 1, José Manuel Aguirre 1
(1) Oral Medicine. Oral & Maxillofacial Pathology Unit. Departmento de Estomatología. Universidad del País Vasco / EHU
(2) Instituto de Investigaciones Odontológicas. Universidad Central de Venezuela
ABSTRACT
Adamantiades-Behçet disease (ABD) is a chronic multisystemic vasculitis that is able to affect any human organ or system.
Recurrent oral ulcers are a very important clinical sign. ABD is a worldwide pathology, which prevalence varies according to the population and geographic location. Although ABD has been known for ages, its aetiology remains an enigma. Genetic, immunological and microbiological factors have been associated. A wide spectrum of clinical manifestations (oral, genital, cutaneous, ocular, neurological, vascular and gastrointestinal) and an unpredictable evolution with repeated periods of exacerbation and remission are the most representative aspects of this pathology. The complex treatment of ABD requires a deep multidisciplinary cooperation; therefore, there is an extensive development of new therapeutic agents that have improved the prognosis of ABD. In this review were analysed the main etiological, clinical and therapeutic aspects of the disease.
Key words: Adamantiades-Behçet disease, Behçet disease, oral ulcers, aphthous stomatitis, vasculitis.
RESUMEN
La enfermedad de Adamantiades-Behçet es una vasculitis multisistémica crónica, potencialmente capaz de afectar a cualquier órgano o sistema del cuerpo humano y en la que la aparición repetida de úlceras orales es una de sus principales expresiones clínicas. La EAB es una patología de carácter universal, con una prevalencia variable en función de la población estudiada y que muestra una curiosa distribución geográfica. A pesar de ser un proceso conocido desde la antigüedad, su etiopatogenia en la que probablemente se hayan implicados factores genéticos, microbiológicos e inmunológicos, continua siendo enigmática. Su amplio espectro de manifestaciones clínicas orales, genitales, cutáneas, oculares, neurológicas, vasculares y gastrointestinales y su impredecible evolución con periodos de exacerbación y de remisión son dos de los aspectos más representativos de esta patología. El complejo tratamiento de la EAB requiere una estrecha cooperación multidisciplinar dado su carácter multisistémico. Gracias a ello y al desarrollo de nuevos agentes terapéuticos su pronóstico ha mejorado sensiblemente con respecto a décadas pasadas. En esta revisión analizamos los principales aspectos etiopatogénicos, clínicos y terapéuticos de esta enfermedad.
Palabras clave: Enfermedad de Adamantiades-Behçet, enfermedad de Behçet, úlceras orales, aftas, vasculitis.
Introduction
Adamantiades-Behçet disease (ABD) is a multisystemic process, which aetiology remains unknown; however it might be produced by an immunological disorder which generates vasculitis (1, 2). Even though ABD might differ from each patient, recurrent oral ulcers are the most frequent manifestation of the disease, however vasculitis potentially involves almost every organ or system of the human body (1-3).
Although ABD was recognised primarily by Hippocrates on the V century BC, there are not any references regarding the diseases until 1931, when the Greek ophthalmologist Benedict Adamantiades described it clinically (1, 4). Later, in 1937, the Turkish dermatologist Hulusi Behçet made a complete description of the disease and established the classical triad "oral, genital and ocular ulcers" (1, 4). Several subsequent investigations allowed relating other manifestations, as cutaneous, joint, digestive, vascular and neurological with the disease.
As a memory of Adamantiadess contribution, some authors have considered (4) the term ABD more appropriated instead of "Behçet disease" only. Also, it is controversial whether the ABD should be considered "a disease" or "a syndrome". Since it is a well described entity some people refer to it as "a disease", however, lack of a knowing aetiology and a wide clinical range seem enough to call it "a syndrome" (1,2).
Epidemiology
ABD is a universal pathology, although it shows a singular geographic distribution. Its prevalence is high in Southeast Asia (2-20/100.000), Mediterranean countries and in the "silk path" countries (13-370/100.000). It is especially frequent in Japan (13-30/100.000), Turkey (80-300/100.000) and South Korea, while its prevalence in low in the rest of the European countries and US (0,1-7,5 casos/100.000) (5-8). Even though certain ethnic groups have shown a higher predisposition for the disease, it is debatable whether it is due to racial or geographic reasons (5-8). ABD afflicts females and males equally in almost all countries, apart from in Middle East countries as Iran, Turkey, Libya, Iraq, Jordania, Israel and Egypt, where studies have shown a higher prevalence among the male group (1.5-5:1) (5-8).
The beginning of the symptoms might appear at any age, however, most of the cases show them around the 3rd decade of life and, usually the disease develops completely after 15 months of first appearance. There are hardly any cases of neonatal cases published and children are scarcely affected and show no clinical differences compared to adults (1-3). An early start of the disease in males is associated with an elevated clinical aggressiveness (9).
Etiopathogenesis
The precise aetiology of ABD is still unknown and differs from the classical patterns of autoimmune diseases. The existence of family cases, the relation to a number of genes and its capricious geographic distribution suggest an association to genetic or environmental factors (1, 10). Nevertheless, since ABD does not follow Mendels laws, the number of familiar cases is very low (1-18%) and it has been impossible to find an exclusive gene alteration, the aetiology of ABD should not be considered solely genetic (1, 10).
It has been demonstrated a narrow relation between ABD and the Major Histocompatibility Complex (MHC) gene HLA-B51, especially in Japan, the Middle East and the Mediterranean countries. This relationship has been observed most commonly in men and in association with ocular symptoms (1-3). Other MHC genes interrelated with ABD are HLA-B27, linked to joint lesions, and HLA-B12, linked to mucocutaneous manifestations (1-3). Nowadays, many investigations are in progress with the intention of connecting alterations of HLA-DRw8, HLA-B15 or MIC-A genes, as well as the adhesion molecule ICAM-1 with the vasculitis process (11-15).
From the first descriptions of ABD, it has been speculated about the relation of the disease to infectious factors, mostly based on the geographical distribution. Interestingly, even Behçet in the original publication, pointed a possible viral aetiology due to the appearance of genital and oral lesions (16). Several studies (1-3) have tried to prove the existence of a viral factor, in particular Herpes family, or a bacterial factor, specifically streptococci, without any luck. Different authors (1-3, 14, 15) have identified DNA of Herpes simplex type 1 from lymphocytes and monocytes, as well as a higher level of antibodies against Herpes simplex type 1 in patients with ABD. However, the proportion of patients with positive viral serology and positive viral DNA from biopsies was not statistically significant. The results of some studies in vitro (1-3, 14, 15) have suggested the possibility that the hypersensibility to certain strains of Streptococcus sanguis might play a relevant role in the etiopathogenesis of the disease. In fact, the frequency of oral lesions might not be casual and be, in some way, related to oral streptococci. For the moment, lack of conclusive evidences forbids from estimating the relevance of infectious agents in ABD.
Another piece of the puzzle represents the immunological alterations observed in patients with ABD. Up to 50% of patients show an increase of circulating immunocomplex during the exacerbation peaks of the disease (1-3, 10). Some studies (10,14,15) have observed during active phases of the disease a decrease of CD4/CD8 lymphocyte ratio at the expense of an increase of the number of CD8+ lymphocytes, a decrease of natural killer (NK) cell activity or an increase of interleukin IL-2, IL-10 or tumour necrosis factor receptor-γ(TNFRγ) levels. Also, it has been observed an increase of γδ lymphocytes able to secrete lymphokines with cytotoxic activity (10, 14, 15). Some of the substances produced by T lymphocytes might play an important role in aphthous stomatitis as fibroblast growth factor (FGF)-7, which stimulates and accelerates fibroblast proliferation, and tumour necrosis factor (TNF), which plays a key role in the pathogenesis of the oral lesions (10-12).
In the absence of a general consensus about the etiopathogenesis, ABD should be considered a multifactorial disease, which cannot be classified isolated as a genetic, autoimmune or infectious disease.
Clinical features
Broad clinical spectrum and unpredictable evolution are the most representative characteristics of ABD (17). Oral, genital, cutaneous, ocular, neurological, vascular and gastrointestinal manifestations are some of the clinical aspects of the disease.
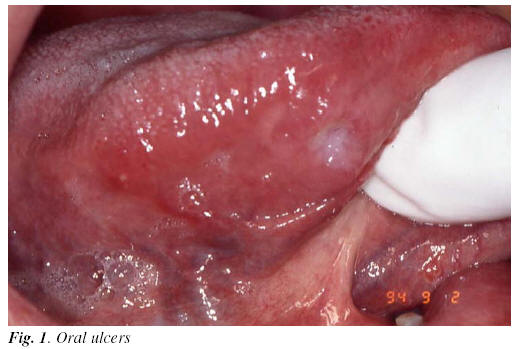
1) Oral lesions: Oral ulcers are found in 95-100 % of the patients (1-3). Clinically, they are indistinguishable from recurrent aphthous stomatitis (RAS). Like RAS, oral ulcers are classified as: minors (< 1 cm.), majors (> 1 cm.) and herpetiform (1-3 mm) (1-17) (Figure 1). There are barely any difference between RAS and ABD. Nevertheless, ABD ulcers tend to appear in unusual places and in a higher number (17). Trauma has been observed as inductive factor in both processes, in addition to the protective role of tobacco, hypothetically based on the hyperkeratosis of the epithelia (17, 18). Oral lesions, in many cases, are the first sign of ABD and, in most of the patients, episodes of oral ulcers persist for 1 to 3 years before appearing the other signs of the disease (17, 18).
2) Genital lesions: Genital lesions are characterised by ulcers also, though they are less frequent than oral lesions (60-80%) (1-3). Ulcers may appear in any place of the genital area, most frequently on the male scrotum and minor and major females lips. Lesions are painful and may cause walk difficulties and even sometimes dysuria. Risk of infection is a common complication during the evolution of the lesions, which increases depending upon the size and depth of ulcers (1-3, 19).
3) Cutaneous lesions: Cutaneous manifestations are observed in 80% of patients with ABD. The most common lesion represents erythema nudosum (40-50%) and pustules or acne-like lesions (60-70%) (20). Lesions erythema nudosum-like are more frequent on inferior limbs, and generally they do not ulcer. After healing they usually leave a hyperpigmented surface. Histologically, a focal vasculitis of small blood vessels is observed (20). Pustules or acne-like lesions may appear at any location and they are morphologically similar to adolescent acne. Histologically, besides vasculitis of small blood vessels, an inflammatory infiltrate compounded from neutrophils and necrosis is observed (20).
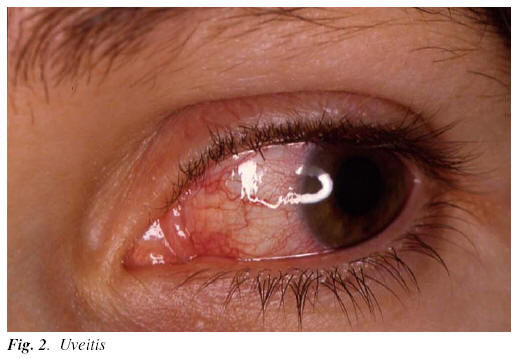
4) Ocular lesions: 20 to 50% of patients with ABD have ocular involvement (1-3). Habitually, they appear two or three years after the beginning of the disease. Manifestations may vary; from a mild conjunctivitis to a vasculitis or retina atrophy, glaucoma, cataracts or retina loosening (17, 21) (Figure 2). The severity of the ocular manifestations is variable and generally bilateral, total or partial lost of the sight is observed in 25% of the patients (21).
5) Joint lesions: Up to 45-50% of patients with ABD may show non deforming arthritis, in one or more joints, which last several days or weeks, mainly affecting limb joints (1-3, 17).
6) Neurological lesions: 5 to 20% of the patients might develop brain or spinal medulla damage or meningitis (22). Clinically, it is manifested by emotional and sensorial alterations, paralysis, paresis, and even severe psychiatric disorders. Cerebrospinal fluid may appear normal or show an increase of neutrophils, lymphocytes or proteins (17, 22).
7) Vascular lesions: Basically, any lesion associated to ABD is produced directly or indirectly by vasculitis. Vasculitis may affect any kind of blood vessels; arteries, veins, deep or superficial; however, small calibre vessels are the most common (23). Tendency for thrombosis is very common, either superficial or deep, particularly in the inferior limbs. Fortunately, complications as lung emboli are rare. For that reason, it is very controversial the use of preventive anticoagulants in patients with ABD (17, 23). The risk of arterial aneurysms and arterial obstruction is higher in ABD patients than healthy individuals. Involvement of major arteries may cause fatal consequences (23).
8) Gastrointestinal lesions: Prevalence of gastrointestinal lesions varies among the population studied. There are particularly frequent amongst Japanese (1-3, 17). Primary manifestations are ulcers, which can appear from the oesophagus to the rectum. Symptoms differ according to location and severity of the lesion (abdominal pain, dysphagia, diarrhoea, etc). Although these lesions are infrequent, complications may be severe (17).
9) Other lesions: There are numerous alterations that have been associated to ABD, however, they seem less frequent than the manifestations described above. The most important are the cardiac complications (pericarditis, coronary thrombosis, valve prolapse), follow by renal (proteinuria, haematuria or glomerulonephritis), pulmonary and pleurae complications (17-23).
Diagnosis
Diagnosis of ABD remains difficult, due to the absence of a conclusive diagnostic test that confirms the clinical diagnosis (1-3). In ancient times "pathergy test" had a great relevance. This test measures the reaction after 24 or 48 hours after an intradermal injection in the forearm (It is considered positive whether an erythematous nodule or pustule larger than 2 mm is formed). Unfortunately the test might result negative in up to 50% of cases (24).
In order to unify inclusion criteria, several groups have established different classifications. The classification produced in 1990 by the International Study Group for Behçets Disease is the most accepted among the international scientific community (25). In this classification is indispensable for the diagnosis 3 episodes of oral ulcers within a 12 month period. Also, patient must have suffered at least 2 of the 4 following manifestations: repeated genital ulcers, ocular lesions, cutaneous lesions or positive pathergy test (25) (Table I). The real applicability of these criteria is for research more than for the clinical practice, as many patients, without filling all the inclusion standards, have clinical features completely consistent to the diagnosis of EAB (24).
As any other disease, or even more due to the absence of diagnostic tests, it is essential to complete a meticulous clinical history in order to establish the appropriate differential diagnoses. Depending on the type of clinical manifestation differential diagnosis should be established with RAS, Neumann aphtous stomatitis, Reiter syndrome, sarcoidosis, multiple sclerosis, Steven-Johnson syndrome, PFAPA syndrome, MAGIC syndrome, Crohn disease or celiac disease.
Treatment
Owing to the multiple organ and tissue involvement of ABD, therapy must be performed for a group of different health professionals. The correct treatment requires a narrow collaboration among different practitioners in order to coordinate an individual protocol for each patient. The therapy always combines a general and an organ-specific approach (26). General therapy controls the active periods of the disease and consists of different anti-inflammatory drugs, steroids or immunotherapy (26). Among the new drugs, use of alpha interferon 2A and B are showing good results. They seem to reduce and prevent symptoms; however, hardly any studies have been done with ABD patients (27).
Regarding to oral lesion treatment, it is basically the same as the one indicated for RAS. Topical treatments are indicated for mild lesions and systemic treatments for severe lesions (28, 29). Among topical therapies, use of steroids is the most popular choice. Triamcinolone (0,05-0,5%), fluocinolone (0,05-0,1 %), clobetasol (0,05-0,1%) and betametasone (0,1%) reduce the symptoms and duration of the ulcers, though they do not prevent recurrences (26-29). The effectiveness of topical drugs, as anaesthetics, antiseptics, and analgesics is limited, thus they should be used as a complementary therapy only (28, 29).
When oral lesions are severe systemic therapies are indicated. Employ of immunodrugs as thalidomide, tacrolimus, azathioprine, azelastine or metotrexate decreases the number of lesions, length and pain, and increases the latency period. However, these drugs produce important secondary effects as teratological anomalies, severe digestive disturbances, haematological problems, etc. For those reasons, indication and use of those drugs must be continuously supervised and women potentially fertile must follow strict programs to prevent conception. Dose is individualised and should be rigorously followed (26-29). Systemic steroids, as prednisone at different dosage, produce rapid heal and longer latency periods, however, its administration must be indicated under strict measures to guarantee minimal side effects (28, 29). Other medicaments like pentoxifiline, an inmunomodulator, or colchicine, an antifungal drug, reduce and prevent new lesions even when the therapy has ended. Their mechanism of action remains unknown (26-29). As the other drugs mentioned above, use of pentoxifiline and colchicine must be indicated under strict surveillance.
Prognosis
ABD is a chronic disease and show surprising periods of remission and activity. Generally, frequency and intensity of the disease decrease with time, though patient evolution is still unpredictable. Depending on the organs and tissues involved, mobility may change. Up to 20% of patients may have a severe evolution and develop several complications as: thrombosis, intestinal perforation, myocardial infarct, aneurisms and others, which could cause death of patient. Fortunately, those cases are rare and preventive therapies reduce the mortality to levels similar to the general population rate (30). Mobility, however, is still very high accompanied with complications as blindness or severe arthritis.
![]() Correspondence
Correspondence
Dr. J.M. Aguirre
Medicina Bucal. Departamento de Estomatología
Facultad de Medicina y Odontología
Universidad del País Vasco EHU
Leioa 48940. Vizcaya.
E-mail: otpagurj@lg.ehu.es
Received: 23-05-2004
Accepted: 30-01-2005
References
1. Kontogiannis V, Powell RJ. Behçet´s disease. Postgrad Med J 2000;76: 629-37. [ Links ]
2. Lee LA. Behcet disease. Semin Cutan Med Surg 2001;20:53-7. [ Links ]
3. Barnes CG, Yazici H. Behcet´s Syndrome. Rheumatology 1999;38:1171-6. [ Links ]
4. Dimakakos PB, Tsiligiris B, Kotsis T. The physician B. Adamantiades and his contribution to the disease Adamantiades-Behcet. Int Angiol 1999; 18:176-81. [ Links ]
5. Zouboulis CC. Epidemiology of Adamantiades-Behçet´s disease. Ann Med Int 1999;150:488-98. [ Links ]
6. Jaber L, Milo G, Halpern GJ, Krause I, Weinberger A. Prevalence of Behçet´s disease in an arab community in Israel. Ann Rheum Dis 2002;61:365-6. [ Links ]
7. Verity DH, Marr JE, Ohno S, Wallace GR, Stanford MR. Behçet´s disease, the silk road and HLA-B51: historical and geographical perspectives. Tissue Antigens 1999;54:213-20. [ Links ]
8. Zouboulis C, Kotter I, Djawari D, Kirch W, Kohl P, Ochsendorf FR et al. Epidemiological features of Adamantiades- Behçet´s disease in Germany and in Europe. Yonsei Med J 1997;38:411-22. [ Links ]
9. Yacizi H, Yurdakul S, Tüzün Y. Influence of age and onset and patient´s sex on the prevalence and severity of manifestations of Behçet´s disease. Ann Rheum Dis 1984;43:783-9. [ Links ]
10. Onder M, Gurer MA. The multiple faces of Behçet´s disease and its aetiological factors. J Eur Acad Dermatol Venereol 2000;15:126-36. [ Links ]
11. Lehner T, Welsh KI, Batchelor JR. The relationship of HLA-B and DR phenotypes to Behçet´s syndrome, recurrent oral ulceration and the class of immune complexes. Immunology 1982;47:581-7. [ Links ]
12. Kim EH, Mok JW, Bang D, Lee ES, Lee S, Park K. Intercellular adhesion molecule-1 polymorphisms in Korea patients with Behçet´s disease. J Korean Med Sci 2003;18:415-8. [ Links ]
13. Sun A, Hsieh RP, Chu CT, Wang JT, Liu BY, Chiang CP. Some specific human leukocyte antigen (HLA)-DR/DQ haplotypes are more important than individual HLA-DR and –DQ phenotypes for the development of mucocutaneous type of Behçet´s disease and for disease shift from recurrent aphthous stomatitis to mucocutaneous type of Behçet´s disease. J Oral Pathol Med 2001;30:402-7. [ Links ]
14. Meador R, Ehrlich G, Von Feldt JM. Behçet´s disease: immunopathologic and therapeutic aspects. Curr Rheumatol Rep 2002;4:47-54. [ Links ]
15. Emmi L, Brugnolo F, Marchione T. Pathogenesis and therapy of Behçet´s disease. Ann Ital Med Int 1997;12:20-5. [ Links ]
16.Behçet H. Uber rezidivierende, aphtose, durch ein virus verursachte. Geschwure am Mund, am Auge und an den Genitalen. Dermatol Wochenchir 1937;36:1152-7. [ Links ]
17.Bang D. Clinical spectrum of Behçet´s disease. J Dermatol 2001;28: 610-3. [ Links ]
18. Rizvi SW, McGrath H Jr. The therapeutic effect of cigarette smoking on oral/genital aphtosis and other manifestations of Behçet´s disease. Clin Exp Rheumatol 2001;19:77-8. [ Links ]
19. Cappele O, Nicolas J, Bottet P, Bensadoum H. Urological manifestations of Behçet´s disease. Prog Urol 2003;77:329-31. [ Links ]
20. Lee E, Bang D, Lee S. Dermatologic manifestations of Behçet´s disease. Yonsei Med J 1997;38:380-9. [ Links ]
21. Tugal-Tutkun I, Onal S, Altan-Yaycioglu R, Huseyin Altunbas H, Urgacioglu M. Uveitis in Behçet disease: An analysis of 880 patients. Am J Ophtalmol 2004;138:373-80. [ Links ]
22. Silva A, Altintas A, Saip S. Behcet´s syndrome and the nervous system. Curr Opin Neurol 2004;17:347-57. [ Links ]
23. Koc Y, Güllü I, Akpek G, Akpolat T, Kansu E, Kiraz S et al. Vascular involvement in Behçet´s disease. J Rheumatol 1992;19:402-10. [ Links ]
24. Kyu H, Cheon KS. The clinical significance of a pathergy reaction in patients with Behçet´s disease. J Korean Med Sci 2002;17:371-4. [ Links ]
25. International Study Group for Behçet´s Disease. Criteria for diagnosis of Behçet´s disease. Lancet 1990;335:1078-80. [ Links ]
26. Whallet AJ, Thurairajan G, Hamburger J, Palmer RG, Murray PI. Behçet´s syndrome: a multidisciplinary approach to clinical care. Q J Med 1999;92: 727-40. [ Links ]
27. Kotter I, Gunaydin I, Zierhut M, Stubiger N. The use of interferon alpha in Behcet disease: review of the literature. Semin Arthritis Rheum 2004; 33:320-35. [ Links ]
28. Sakane T, Takeno M. Current therapy in Behçet´s disease. Skin Therapy Lett 2000;5:3-5. [ Links ]
29. Barrons RW. Treatment strategies for recurrent oral aphtous ulcers. Am J Health Syst Pharm 2001;58:41-53. [ Links ]
30. Yacizi H, Basaran G, Hamuryudam V, Hizli N, Yurdakul S, Mat C et al. The ten-year mortality in Behçet´s syndrome. Br J Rheumatol 1996;35:139-41. [ Links ]

