










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroducción
En los últimos años hemos asistido a una oferta creciente de tratamientos de medicina regenerativa por parte de numerosos establecimientos o clínicas, en especial de medicina deportiva, cirugía ortopédica y traumatología, medicina estética, dermatología, odontología, cirugía plástica, cirugía maxilofacial y oftalmología, gracias a la introducción en el mercado de diversos dispositivos de fácil manejo que, a partir de sangre, células o tejidos de los propios pacientes, permiten la elaboración de productos biológicos autólogos y su aplicación dentro de un mismo procedimiento1-3. Estas terapias, basadas en concentrados de plaquetas o de células, habitualmente denominadas «madre», se ofrecen como tratamientos innovadores y se anuncian, a pesar de estar prohibida su publicidad4,5, a modo de marchamo de calidad de establecimientos que buscan diferenciarse de sus competidores6. No obstante, más allá de las dudosas eficacia y seguridad de muchos de los tratamientos que se ofrecen bajo el reclamo de las células madre o de la medicina regenerativa, que muchas veces conllevan riesgos para la salud infravalorados7, la mayor parte de los centros que ofrecen estos tratamientos desconocen los requisitos y las implicaciones legales de su uso. El principal error radica en confundir las autorizaciones que requiere el propio dispositivo con las que requiere el producto obtenido para su aplicación en pacientes5; confusión propiciada, en ocasiones, por las propias empresas que comercializan los equipamientos. Una práctica observada en empresas del sector es publicitar que sus dispositivos disponen de todas las autorizaciones necesarias, cuando en realidad la autorización solo se refiere al equipamiento, no al uso del producto obtenido8, y se traslada una falsa seguridad a los profesionales involucrados en la indicación y la administración de los productos habitualmente sin la autorización requerida, quienes asumen responsabilidades muchas veces desconocidas.
En este artículo se presentan, de forma resumida, los principales requisitos para el uso de estos productos biológicos autólogos obtenidos mediante diversos dispositivos con el objetivo de que puedan servir de guía tanto para los profesionales que los prescriben como para aquellos que inspeccionan los centros donde se administran. También se ofrecen algunas recomendaciones para los pacientes. El ámbito de revisión se limita a la principal regulación europea y española vigente de aplicación a productos celulares y plasmáticos autólogos, específicamente los medicamentos de terapia avanzada, los trasplantes de células y tejidos, y el plasma rico en plaquetas (PRP), incluyendo los documentos con carácter de consideraciones, notas informativas e informes de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) y la Organización Nacional de Trasplantes.
Consideración legal de los dispositivos y de los productos obtenidos
Los dispositivos utilizados para la elaboración de productos biológicos autólogos tienen la consideración de productos sanitarios y, como tales, requieren disponer del correspondiente marcado CE otorgado por un Organismo Notificado dentro de la Unión Europea. Dicho marcado CE solo certifica un mecanismo de producción, no el producto final, y no implica la autorización del uso del producto obtenido4,5.
Respecto a los productos obtenidos, se distinguen fundamentalmente dos tipos: el PRP y los concentrados celulares. El PRP se obtiene a partir de la sangre del paciente y, dependiendo del protocolo de producción empleado, tiene una concentración variable de plaquetas, leucocitos, eritrocitos y moléculas bioactivas, y puede contener diferentes factores de crecimiento, citocinas y proteínas implicadas en la adhesión celular (fibrina, fibronectina y vitronectina). Bajo la nomenclatura de PRP se engloban el preparado rico en factores de crecimiento (PRGF), el plasma rico en plaquetas y factores de crecimiento (PRPGF), el plasma rico en plaquetas (PRP), el plasma pobre en plaquetas (PPP), el plasma rico en plaquetas y rico en leucocitos (LR-PRP), y el plasma rico en plaquetas y pobre en leucocitos (LP-PRP). El PRF (fibrina rica en plaquetas) se considera una segunda generación de PRP que también contiene leucocitos, citocinas y factores de crecimiento, pero dentro de una matriz tridimensional de fibrina. El PRF, cuando es autólogo, se considera medicamento de uso humano de fabricación no industrial para «atender necesidades especiales»4.
Los concentrados de células progenitoras o células troncales (denominadas coloquialmente «células madre») son productos que pueden aislarse a partir de diferentes tejidos, entre ellos la médula ósea, la sangre periférica y otros de fácil acceso, como la dermis obtenida a partir de una biopsia o sacabocados de piel, o la fracción vascular estromal que se obtiene de tejido adiposo previamente lipoaspirado. La composición celular de estos productos es variable y, según el tipo de manipulación y el uso que quiera darse al producto, puede tratarse de células para trasplante o, con más frecuencia, de medicamentos de terapia avanzada5, que están sometidos a una estricta regulación9. El producto celular se considera medicamento de terapia avanzada si las células o tejidos han sufrido una modificación sustancial10,11. No se consideran modificaciones sustanciales el corte, la trituración, el moldeo, la centrifugación, la imbibición en disoluciones antibióticas o antimicrobianas, la esterilización, la irradiación, la separación, concentración o purificación celular, el filtrado, la liofilización, la congelación, la criopreservación o preservación, ni la vitrificación10. Especialmente relevante es que también se considera medicamento de terapia avanzada si las células, aun no habiendo sido modificadas de forma sustancial, se utilizan buscando una función que difiere de las suyas esenciales10,11. Por tanto, aun en el caso de sistemas de obtención celular que no conllevan manipulación sustancial, si el producto celular se utiliza para una función diferente de la suya originaria, se trataría de un medicamento de terapia avanzada. Este es el caso, por ejemplo, de las células obtenidas de tejido adiposo, de mucosa o de piel que se utilizan con finalidad regenerativa de otros tejidos, como hueso, tendones, ligamentos o cartílago. La Agencia Europea de Medicamentos (EMA) se ha pronunciado en reiteradas ocasiones clasificando este tipo de productos como medicamento de terapia avanzada12. En cambio, la obtención de tejido o de células de una zona del cuerpo con el objetivo único de aplicarlo en otra zona diferente para su misma función no se consideraría un medicamento de terapia avanzada; este sería el caso, por ejemplo, de la obtención de tejido adiposo de una zona del cuerpo para proceder al relleno de otra zona diferente, como en los procedimientos de lipoescultura.
Métodos de obtención de PRP y concentrados celulares autólogos. Principales requisitos de calidad
El crecimiento experimentado en la oferta de terapias regenerativas se debe fundamentalmente a la actual disponibilidad de diversos dispositivos comerciales que, como ya se ha comentado, deben disponer del correspondiente marcado CE. En general, estos dispositivos consisten en sistemas cerrados semiautomatizados.
También existe la posibilidad de obtener concentrados celulares y PRP mediante métodos manuales que implican sistemas denominados «abiertos», cuyos requisitos de calidad varían en función del tipo de producto (Fig. 1). En el caso del PRP, el procesamiento tiene que hacerse de acuerdo con las Normas de Correcta Fabricación de Medicamentos y en laboratorios que han de ser inspeccionados por la autoridad competente de las comunidades autónomas13,14. Los medicamentos de terapia avanzada deben fabricarse en laboratorios que cumplan con las Normas de Correcta Fabricación específicas de medicamentos de terapia avanzada15. Finalmente, en el caso de los productos considerados como trasplante, el procesamiento tiene que hacerse en establecimientos de tejidos autorizados por las autoridades sanitarias de las comunidades autónomas de acuerdo con los requisitos establecidos por la legislación española16.
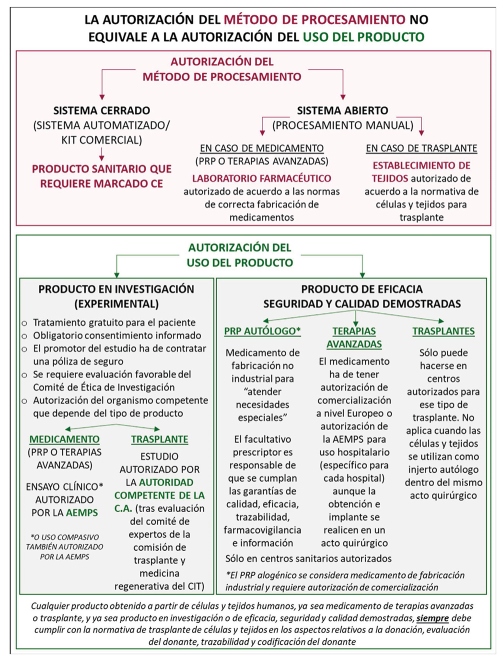
Figura 1. Aspectos fundamentales de la autorización de los métodos de procesamiento y del uso de productos biológicos autólogos en medicina regenerativa. AEMPS: Agencia Española de Medicamentos y Productos Sanitarios; CA: comunidad autónoma; CIT: Consejo Interterritorial del Sistema Nacional de Salud; PRP: plasma rico en plaquetas (denominación genérica que engloba diversos productos, incluida la fibrina rica en plaquetas, que varían en el contenido de distintos tipos de células, citocinas y factores de crecimiento).
Principales implicaciones y requisitos para el uso de PRP
Con independencia del método de obtención del PRP, su uso se regula de igual manera, de acuerdo con lo establecido por la AEMPS4,13,14. Este tipo de medicamentos solo pueden ser prescritos por médicos, odontólogos o podólogos, en el ámbito de sus competencias respectivas, con la cualificación adecuada, con experiencia en el tratamiento, con el equipamiento o instrumentación adecuada, y en establecimientos y centros sanitarios que estén debidamente autorizados de acuerdo con la normativa vigente en las respectivas comunidades autónomas. Además, como cualquier otro medicamento sujeto a prescripción médica, queda prohibido cualquier tipo de publicidad destinada al público en general.
Es especialmente relevante saber que el prescriptor es el responsable de garantizar el cumplimiento de las garantías de calidad antes mencionadas (aunque el procesado y la obtención los realice otro). El facultativo prescriptor también es responsable de que se cumplan el resto de las garantías que exige la regulación, como las de trazabilidad, farmacovigilancia e información, así como las garantías de eficacia, es decir, que se va a utilizar el PRP para indicaciones en las que existe suficiente evidencia científica sobre su eficacia. La propia AEMPS subraya que en pocas indicaciones se han realizado ensayos clínicos de buena calidad. Por otra parte, la AEMPS informa de que, en colaboración con expertos y con las principales sociedades científicas implicadas, establecerá un listado de aplicaciones sobre las que existe evidencia de un balance beneficio-riesgo favorable al uso de cada PRP y aquellas en las que será necesario realizar ensayos clínicos para aceptar dicho uso, si bien hasta la fecha no se ha publicado ese listado. No obstante, recientemente el Consejo de Europa ha publicado la cuarta edición de su guía de calidad y seguridad de tejidos y células para uso humano, en la que se señala, respecto al uso del PRP, la falta de evidencia científica obtenida a partir de ensayos clínicos controlados y aleatorizados a gran escala, y se indica la necesidad de su realización para demostrar la eficacia17. Por ello, antes de poner en marcha la técnica para una determinada indicación, recomendamos consultar con la AEMPS.
Principales implicaciones y requisitos para el uso de concentrados celulares autólogos
En el caso de los medicamentos de terapia avanzada, la normativa que los regula se aplica en todos los casos, aunque sean medicamentos obtenidos con dispositivos que permiten su utilización dentro de un procedimiento quirúrgico (punto 2.47 de las normas de correcta fabricación de medicamentos de terapia avanzada)15. Es imprescindible tener presentes las consideraciones publicadas por la AEMPS en julio de 2018 sobre los productos sanitarios utilizados para la obtención de células autólogas y la clasificación del producto resultante como medicamento de terapia avanzada5. Dado que actualmente ningún medicamento de terapia avanzada obtenido mediante dispositivos o kits (que permiten su uso dentro de un procedimiento quirúrgico) dispone de autorización de comercialización18,19 (que solo puede obtenerse a través de la EMA10) ni de autorización de uso hospitalario20,21 (que solo puede obtenerse de la AEMPS), todos estos tratamientos se consideran medicamentos en investigación que únicamente pueden ser ofrecidos a los pacientes de forma gratuita y dentro de un ensayo clínico autorizado por la AEMPS22, o como uso compasivo23 que también debe ser autorizado por la AEMPS. Además, es obligatorio informar al paciente del carácter experimental del tratamiento y recabar su consentimiento.
Por otra parte, en el caso de trasplantes de células y tejidos que tengan consideración de terapias experimentales, el promotor, además de remitir el estudio para su evaluación al comité de ética de la investigación correspondiente, tiene que solicitar la autorización a la autoridad competente de su comunidad autónoma (habitualmente la Coordinación Autonómica de Trasplantes), que ha de requerir su evaluación por el comité de expertos de la Comisión de Trasplantes y Medicina Regenerativa del Consejo Interterritorial del Sistema Nacional de Salud24.
Finalmente, los medicamentos de terapia avanzada fabricados a partir de células y tejidos humanos, sean autólogos o no, y tanto medicamentos en investigación como autorizados para su uso hospitalario o para su comercialización, siempre deben cumplir con la normativa de trasplante de células y tejidos en los aspectos relativos a donación, evaluación del donante, trazabilidad y codificación del donante9. Por tanto, al igual que en los trasplantes, solo pueden obtenerse células y tejidos en centros debidamente autorizados para ello por las autoridades sanitarias de la comunidad autónoma de acuerdo con los requisitos establecidos en la legislación española16.
Algunas conclusiones y recomendaciones
El uso de productos biológicos autólogos basados en plasma o en células de los propios pacientes está cada vez más extendido, y se ofrecen como tratamientos de medicina regenerativa sencillos y rápidos. No obstante, a pesar de que España dispone de una regulación de las más avanzadas del mundo y de los esfuerzos de los organismos reguladores por difundir los requisitos para su adecuada aplicación en los pacientes, muchas veces estos tratamientos se aplican sin las suficientes garantías o incluso fuera de la legalidad.
Es preocupante en especial la utilización de medicamentos, ya sean de terapia avanzada o no, que aún no disponen de las suficientes garantías de calidad, seguridad y eficacia, y que por tanto deberían ofrecerse exclusivamente dentro de un ensayo clínico autorizado, tras recabar el consentimiento informado del paciente para someterse a un tratamiento experimental y de forma gratuita. Estas prácticas podrían suponer tanto un fraude para el paciente como un riesgo para su salud.
Por todo ello, es importante el esfuerzo que diversas sociedades científicas y organismos reguladores estamos haciendo por difundir los requisitos para la adecuada aplicación de estos tratamientos entre profesionales, inspectores sanitarios y pacientes. Estos últimos, antes de someterse a un tratamiento con sus propias células fuera de un ensayo clínico y pagar por ello, deberían solicitar a quien se lo prescribe la correspondiente autorización para el uso hospitalario del producto (de la AEMPS) o para su comercialización (de la Comisión Europea). Si el prescriptor afirma que se trata de un tratamiento de medicina regenerativa no considerado medicamento, el paciente debería preguntar al profesional que se lo prescribe por el pronunciamiento de la AEMPS al respecto, dado que es el organismo al que corresponde establecer dicha consideración5. En caso de dudas, siempre es recomendable contactar directamente con la propia AEMPS o con la Organización Nacional de Trasplantes.

