










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versión impresa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.80 no.4 abr. 2005
COMUNICACIÓN CORTA
SÍNDROME DE BARDET-BIEDL
BARDET-BIEDL SYNDROME
MARTÍNEZ GIRALT O1, FLORES DESPRADEL I2
| RESUMEN Se presenta un caso de síndrome de Bardet-Biedl. Palabras clave: Retinosis pigmentaria, obesidad, retraso mental y polidactilia. | SUMMARY We report a case of Bardet-Biedl syndrome. Key words: Retinal dystrophy, obesity, mental retardation, polydactyly. |
Recibido: 28/5/04. Aceptado: 12/4/05.
1 Doctora en Medicina. Hospital de l'Esperança del Mar. UAB. Barcelona.
2 Licenciada en Medicina. Hospital de Niños de Barcelona.
Comunicación presentada como póster en el LXXIX Congreso de la S.E.O. (Valencia 2003).
Correspondencia:
Olga Martínez Giralt
Servicio de Oftalmología
Hospital de l'Esperança
Sant Josep de la Muntanya, 12
08024 Barcelona
España
E-mail: olgamgiralt@hotmail.com
INTRODUCCIÓN
Se aporta un caso de síndrome de Bardet-Biedl (SBB). El síndrome de Bardet-Biedl es una afección autosómica recesiva caracterizada por la presentación de distrofia retiniana, obesidad, polidactilia, retraso mental e hipogonadismo (1,2). Las alteraciones cardíacas y renales también son frecuentes.
CASO CLÍNICO
Se presenta el caso de un niño, explorado desde la edad de 4 años. Como antecedentes presenta obesidad, dificultad de escolarización debido al discreto retraso psicomotor, alteración de la dentición (fig. 1), polidactilia en los dedos de los pies intervenida a las siete semanas (figs. 2 y 3) y sin alteración en las manos (fig. 4) y defecto refractivo (astigmatismo miópico) corregido con graduación óptica. Al cabo de cinco años acude de nuevo a control por problemas de adaptación a la oscuridad y disminución de la agudeza visual (de 0,7 a 0,4 en ambos ojos). A la exploración del fondo de ojo se detecta palidez papilar y mínimos cambios en el epitelio pigmentario a nivel del ecuador. Se practican pruebas electrofisiológicas oculares de potencial evocado visual y electrorretinograma. La conducción occipital evocada era normal.

Fig. 1. Alteraciones dentales y obesidad.
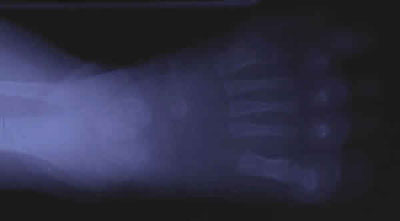
Fig. 2a. Radiología del pie derecho: polidactilia.
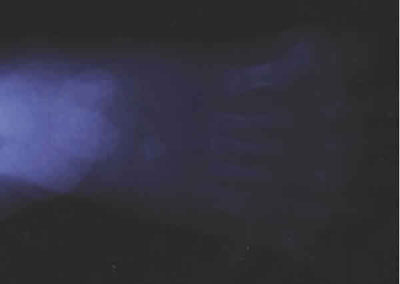
Fig. 2b. Radiología del pie izquierdo: polidactilia.
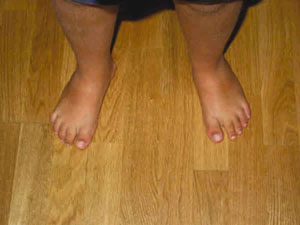
Fig. 3. Polidactilia intervenida a las siete semanas de edad.
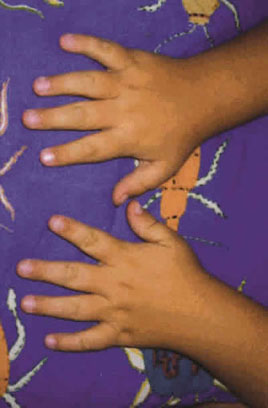
Fig. 4. Anatomía de las manos normal.
La respuesta escotópica y fotópica estaban abolidas (fig. 5). Las exploraciones neurológica y sistémica eran normales. Ante la asociación de distrofia retiniana de bastones y conos, obesidad, polidactilia, retraso mental y alteraciones dentales se diagnosticó de síndrome de Bardet-Biedl.
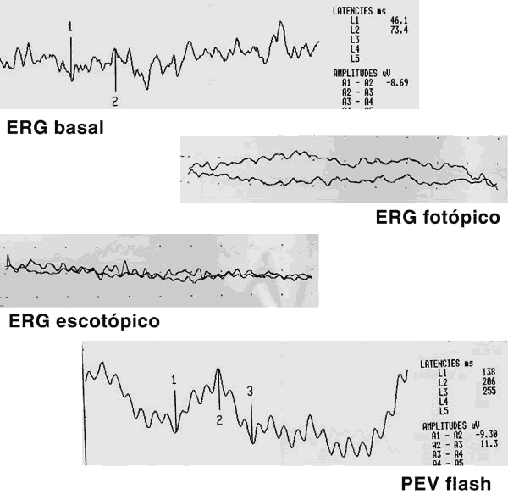
Fig. 5. Pruebas electrofisiológicas: ERG fotópico y escotópico abolidos
y respuesta occipital evocada disminuida.
DISCUSIÓN
El síndrome de Bardet-Biedl (SBB) es un trastorno autosómico recesivo. A pesar de que los estudios genéticos han revelado cinco formas distintas del SBB relacionados con diferentes locus en distintos cromosomas el diagnóstico de SBB se sigue haciendo sobretodo en base a datos clínicos (1). Las manifestaciones clásicas incluyen retinosis pigmentaria (RP), obesidad, polidactilia, hipogonadismo en los niños y retraso mental moderado (2). El caso que se presenta tiene a nivel oftalmológico discreta palidez papilar y mínimos cambios de epitelio pigmentado a nivel ecuatorial con disminución de la agudeza visual y problemas de adaptación a la oscuridad. En general, los exámenes oftalmoscópicos y electrofisiológicos son esenciales para confirmar el diagnóstico de SBB (3). Las pruebas electrofisiológicas practicadas demuestran un potencial evocado visual (PEV) conservado, electrorretinograma (ERG) y amplitud disminuida, electrorretinograma (ERG) escotópico y fotópico abolidos. En un estudio realizado sobre 13 pacientes pediátricos (4) Spaggiari E et al, encontraron una disminución progresiva de la agudeza visual de manera precoz, en la primera década de la vida, signos de retinosis pigmentaria sólo en el 46% de los niños, grandes anomalías en el electrorretinograma de todos los pacientes, incluso en los que no presentaban RP, estos resultados electrorretinográficos, cuando se detectaron sugirieron mayor afectación del sistema fotópico. Como anomalías ortopédicas (5) puede encontrar polidactilia postaxial, escoliosis, tibia valga, tibia vara y otros trastornos, en nuestro caso sólo se apreció la polidactilia en los pies.
La expresión del SBB varía dentro de la misma familia y entre familias por lo que a menudo el diagnóstico es difícil. La edad media del diagnóstico estaba en 9 años (2), lo cual es tarde para esta afección debilitante, pero el lento desarrollo de los aspectos clínicos probablemente contribuyen a ello. La polidactilia postaxial ya está presente en el 69% de los pacientes en el momento de nacer (2), pero la obesidad empieza a desarrollarse alrededor de 2-3 años y la degeneración retiniana no empieza a ser aparente hasta una edad media de 8,5 años. Como nuevos criterios clínicos se incluyen aspectos neurológicos, problemas del habla y lenguaje, alteraciones de conducta, dismorfismo facial y anomalías dentales (2). Incluyendo estos factores dentro de los criterios diagnósticos se podría facilitar un diagnóstico precoz de este trastorno. La evidencia de un fenotipo muy superpuesto al síndrome de Laurence-Moon ha llevado a una propuesta de unificación, con una descripción de síndrome polidactilia-obesidad y alteraciones reno- ocular.
BIBLIOGRAFÍA
1. Carmi R, Rokhlina T, Kwitek-Black AE, Elbedour K, Nishimure D, Stone EM. Use of a DNA pooling strategy to identify a human obesity syndrome locus on chromosome 15. Hum Mol Genet 1995; 4: 9-13. [ Links ]
2. Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet 1999; 36: 437-446. [ Links ]
3. Ingster-Moati I, Rigaudiere F, Choltus-De Petigny MC, Bremond-Gignac D, Lestrade C, Grall Y. Functional visual explorations of Bardet-Biedl syndrome. A study of three cases. J Fr Ophtalmol 2000; 23: 802-808. [ Links ]
4. Spaggiari E, Salati R, Nicolini P, Borgatti R, Pozzoli U, Polenghi F. Evolution of ocular clinical and electrophysiological findins in pediatric Bardet-Biedl syndrome. Int Ophthalmol 1999; 23: 61-67. [ Links ]
5. Ramírez N, Marrero L, Carlo S, Cornier AS. Orthopaedic manisfestations of Bardet-Biedl syndrome. J Pedriat Orthop 2004; 24: 92-96. [ Links ]

