










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versión impresa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.84 no.1 ene. 2009
Tumores de la conjuntiva
Conjunctival tumors
Saornil M.A.1, Becerra E.2, Méndez M.C.3, Blanco G.4
Unidad de Oncología Ocular del CSUR del Sistema Nacional de Salud. Hospital Clínico Universitario de Valladolid.
Registro de Patología Ocular Miguel N Burnier. Instituto de Oftalmobiología Aplicada. Universidad de Valladolid. España. En el momento de la redacción del artículo, todos los autores formaban parte del Registro de Patología Ocular Miguel N Burnier.
1 Doctora en Medicina. Hospital Clínico Universitario de Valladolid. Valladolid. España.
2 Licenciado en Medicina. Becario de la Fundación Carolina en el IOBA 2005. Valladolid. España.
3 Doctora en Medicina. Departamento de Anatomía Patológica de la Universidad de Valladolid. Valladolid. España.
4 Doctor en Medicina. Hospital Universitario del Río Hortega. Valladolid. España.
Dirección para correspondencia
RESUMEN
Los tumores de la conjuntiva son unos de los más frecuentes del ojo y anejos. Abarcan un amplio espectro desde lesiones benignas como el papiloma a otras malignas que pueden poner en peligro la función visual y la vida del paciente, como el carcinoma epidermoide y el melanoma. Pueden surgir de cualquiera de las células que componen la conjuntiva aunque los más frecuentes son los de origen epitelial y melanocítico. El diagnóstico precoz es fundamental para prevenir la extensión ocular y sistémica y para preservar la función visual. En este artículo se revisan las características clínicas de los tumores conjuntivales más frecuentes y se discute su tratamiento.
Palabras clave: Tumores conjuntivales, neoplasia intraepitelial, nevus, melanoma, linfoma.
ABSTRACT
Conjunctival tumors are one of the most frequent of the eye and adnexa. They comprise a large variety of conditions, from benign lesions such as nevus or papiloma, to malignant lesions such as epidermoid carcinoma or melanoma which may threaten visual function and the life of the patient. They can arise from any cellular component, but the most frequent are of epithelial and melanocytic origin. Early diagnosis is essential for preventing ocular and systemic spread and to preserve visual function. In this paper we review the clinical characteristics of the most frequent conjunctival tumors, and we discuss tumor management (Arch Soc Esp Oftalmol 2009; 84: 7-22).
Key words: Conjunctival tumors, intraepithelial neoplasia, nevus, melanoma, lymphoma.
Epidemiología y clasificación
Los tumores de la conjuntiva son los más frecuentes del ojo y anejos junto con los de los párpados. Abarcan un amplio espectro desde lesiones benignas como el papiloma a otras que pueden poner en peligro la función visual y la vida del paciente, como el carcinoma epidermoide y el melanoma si no son diagnosticadas precozmente (tabla I) (1,2). Pueden surgir de cualquiera de las células que componen la conjuntiva aunque los mas frecuentes son los de origen epitelial y melanocítico (tabla I). Los tumores epiteliales son entre un tercio y la mitad del total, siendo la prevalencia más alta en los países con mayor exposición actínica. Con respecto a los tumores melanocíticos, la mayoría son benignos, reconociéndose variaciones según la pigmentación racial y la edad de los pacientes.1,2 En la unidad de Oncología Ocular del Hospital Clínico Universitario de Valladolid, entre 1993 y 2007 se han diagnosticado 314 tumores conjuntivales, de los cuales 149 (48%) fueron melanocíticos (87% benignos), 124 (39,6%) de origen epitelial (64,5% precancerosos), 30 (9,6%) originados en tejidos blandos y 10 (3,2%) de estirpe linfoid (3,4). En la mayoría de los casos, la diferenciación clínica entre lesiones benignas precancerosas y malignas es difícil, precisando la realización de una biopsia para el diagnóstico definitivo. En muchas ocasiones la biopsia, es terapéutica al mismo tiempo ya que conlleva la extirpación completa en el caso de lesiones circunscritas (biopsia escisional).

Recuerdo anatomo-histológico
La conjuntiva es una membrana mucosa fina y flexible que se extiende desde la superficie interna de los párpados (palpebral) a los fornix y a la superficie anterior del globo ocular (bulbar) hasta el limbo esclero-corneal (limbal). Entre sus funciones se encuentran la contribución al film lagrimal precorneal mediante la producción de la capa mucosa, el ser una importante barrera para los cuerpos extraños y las infecciones y la zona límbica en el mantenimiento del epitelio corneal. La vascularización la recibe de ramas de las arcadas marginales de los párpados (tarsal) y de las arterias ciliares anteriores (bulbar). Las conexiones linfáticas de la conjuntiva son paralelas a las de los párpados drenando a los nódulos linfáticos preauriculares y submandibulares y la inervación sensorial la recibe del V par craneal (1,2,5,6).
Histológicamente (5,6) es similar a otras membranas mucosas y se compone de un epitelio estratificado no queratinizado de dos o más capas sobre un estroma formado por tejido conectivo fibrovascular que contiene vasos, nervios y linfáticos (fig. 1). En la capa basal del epitelio se observan melanocitos que producen melanina y la inyectan a las células de alrededor. A lo largo de todo el epitelio se pueden observar células caliciformes, encargadas de producir el componente mucoide del film lagrimal, que son más numerosas en la porción ínfero nasal de la conjuntiva bulbar. En el canto interno se encuentran el pliegue semilunar y la carúncula en la que se pueden encontrar folículos pilosos finos y glándulas sebáceas e incluso glándulas lagrimales accesorias.

Fig. 1: Histología de conjuntiva normal: Epitelio estratificado
no queratinizado con melanocitos en la capa basal y células
caliciformes que tiñen positivamente con la tinción de PAS (40x).
A lo largo de la conjuntiva bulbar y hasta los pliegues subtarsales de los parpados existe una capa linfoide que en algunas zonas forma agregados especializados llamados Tejido Linfoide Asociado a Conjuntiva (Conjunctival-associated Lymphoid Tissue -CALT) correspondiente al Tejido Linfoide Asociado a Mucosas (Mucosa-associated Lymphoid Tissue -MALT) de otras regiones del organismo, formados por acúmulos de linfocitos T y B relacionados con el proceso de información antigénica (5-7).
Tumores epiteliales
Benignos
Papiloma escamoso. Se presentan como lesiones exofíticas, de color sonrosado, de consistencia blanda y superficie irregular. Pueden ser de etiología viral (papilomavirus humano), y en este caso pueden recurrir. Tienden a ser pedunculados en niños (más frecuentes en fórnix inferior) y sesiles (mas frecuentes en conjuntiva bulbar) en adultos. Suelen ser asintomáticos, sin signos inflamatorios asociados. Clínicamente pueden ser difíciles de distinguir de lesiones precancerosas y carcinomatosas. Histológicamente consisten en ejes conectivo-vasculares recubiertos de epitelio conjuntival acantótico, sin signos de atipia (fig. 2) (1,2,8).

Fig. 2: Papiloma escamoso: A: Lesión sesil en conjuntiva bulbar,
sonrosada, ricamente vascularizada. B: Histopatológicamente se
observa epitelio acantótico alrededor de ejes conectivo-vasculares
(Hematoxilina eosina HE4x).
Hiperplasia pseudoepiteliomatosa o pseudocarcinomatosa. Es una hiperplasia del epitelio reactiva a inflamación o procesos de irritación crónica. La fuente de la inflamación suele ser una pinguécula o pterigion preexistente con inflamación crónica en los que el epitelio comienza a proliferar y queratinizarse. Clínicamente consiste en una masa elevada de color sonrosado, de crecimiento rápido, similar a una lesión carcinomatosa, que puede tener aspecto leucopláquico. Histológicamente el epitelio prolifera formando lóbulos, y presenta un aumento de actividad mitótica secundario a la inflamación; se distingue del carcinoma escamoso por la falta de atipia nuclear y la marcada inflamación estromal subyacente. Una variante de esta lesión con morfología redondeada, bordes abruptos y elevados es el queratoacantoma (1,2).
Precancerosos
Queratosis actínica. Son lesiones leucopláquicas, bien circunscritas, elevadas, límbicas, que crecen lentamente en el epitelio del área interpalpebral, generalmente sobre una pinguécula o pterigion preexistente y pueden simular un carcinoma. Histológicamente se caracterizan por placas de epitelio acantótico e hiperqueratótico de bordes abruptos con diferentes grados de atipia celular que no suele alcanzar la totalidad del epitelio (displasia leve o moderada), y degeneración elastótica en el estroma. Puede evolucionar a carcinoma escamoso de conjuntiva, aunque es raro (fig. 3) (1,9).

Fig. 3: Queratosis Actínica. A: Lesión límbica, elevada, leucopláquica.
B: Epitelio acantótico, con una displasia leve y queratinización
superficial que confiere el aspecto leucopláquico (HE4x).
Neoplasias intraepiteliales: Displasias/carcinoma in situ. Es uno de los tumores mas frecuentes de la superficie ocular con una incidencia aproximada de 2 casos/100.000 personas por año. Clínicamente aparece en pacientes de 60-70 años de piel clara. Pueden aparecer en jóvenes inmunosuprimidos y pueden ser bilaterales. Son lesiones gelatinosas, sesiles o papilomatosas, con tendencia a la extensión difusa superficial, generalmente en la hendidura interpalpebral afectando al limbo con bordes mal definidos y suelen extenderse sobre el epitelio corneal. Patogénicamente son afines a las displasias que aparecen en las mucosas no expuestas, y en su desarrollo se ha implicado el papilomavirus humano (mas aun en pacientes con enfermedad bilateral e inmunosuprimidos), el virus de la inmunodeficiencia adquirida, la exposición solar y factores ambientales como exposición a derivados del petróleo, aceites industriales, tabaquismo etc. (fig. 4) (2,10,11).

Fig. 4: Neoplasias Intraepiteliales Conjuntivales circunscritas:
lesiones límbicas, exofíticas, vascularizadas con límites netos, que invaden córnea.
La apariencia clínica es consecuencia de los cambios histológicos que consisten en la aparición de una hiperplasia epitelial de grado variable, en la que parte o todo el espesor del epitelio corneal se sustituye por una proliferación células atípicas (displasia) que comienza en las capas basales del epitelio pudiendo afectar a parte (displasia leve, moderada o grave) o la totalidad del epitelio (carcinoma in situ). El termino CIN (conjunctival intraepithelial neoplasia), incluye los distintos grados de displasia (leve, moderada y grave) (fig. 5) y el carcinoma in situ (fig. 6), implicando que estas entidades son diferentes niveles del espectro del desarrollo de una neoplasia intraepitelial, considerada como lesión precancerosa, ya que si las células atípicas rompen la membrana basal e invaden el tejido subconjuntival se produce el carcinoma escamoso invasivo de conjuntiva (1,9,10). El riesgo de desarrollar carcinoma escamoso es bajo, pero probablemente mayor que el de la queratosis actínica.

Fig. 5: Neoplasia intraepitelial conjuntival con displasia grave que
se extiende más de dos tercios del espesor del epitelio pero
respeta las capas superficiales y la membrana basal (HE40x).

Fig. 6: Neoplasia Intraepitelial Conjuntival Difusa. A: Lesión conjuntival difusa,
elevada y vascularizada que cubre toda la superficie corneal. B: Carcinoma
in situ de conjuntiva. Hiperplasia epitelial con displasia que se extiende
a todas las capas del epitelio (HE10x).
Debido a la imprecisión de sus limites, y a la tendencia al crecimiento difuso, es mas fácil la resección incompleta que en la queratosis actínica, dando lugar a recidivas frecuentes. Su curso, comparado con lesiones similares en otras localizaciones de la economía, es relativamente benigno, ya que suele permanecer confinado al epitelio, y raras veces se convierte en invasivo. Es decir, la mayoría de estas lesiones no sigue el curso de un verdadero tumor maligno, invasivo y metastatizante creando un mayor problema la extensión superficial y las recurrencias que el riesgo de extensión sistémica. El diagnóstico precoz es importante porque la biopsia escisional con margen de resección con o sin tratamientos adyuvantes suele ser un tratamiento definitivo. Sin embargo lesiones avanzadas pueden extenderse por la totalidad de la cornea y gran parte de la conjuntiva haciendo difícil la extirpación completa, poniendo en peligro la función visual y precisando tratamientos quirúrgicos complejos y terapias adyuvantes como quimioterapia tópica (Mitomicina C, 5-Fluorouracilo, Interferon alfa-2b) (12-17) (fig. 7).

Fig. 7: Neoplasia Intraepitelial Corneoconjuntival difusa de >1 año
de evolución (A) y post tratamiento con extirpación parcial-biopsia
de 2/3 de la lesión y tratamiento de la masa residual con Mitomicina C tópica (B).
Malignos
Carcinoma escamoso. Se originan a partir de las queratosis actínicas y neoplasias intraepiteliales y aparecen cuando un carcinoma in situ rompe la membrana basal e invade el tejido subconjuntival accediendo a los vasos linfáticos y adquiriendo potencial metastático. Clínicamente aparece como una lesión exofitica, sesil o pedunculada en el área de exposición interpalpebral, de aspecto variable, frecuentemente cerca del limbo, de crecimiento lento. En su evolución natural, puede crecer ocupando toda la conjuntiva bulbar y extenderse posteriormente a través del septum orbitario, invadiendo la orbita (fig. 8), o invadir la lamela esclerocorneal penetrando en el globo ocular (18,19). Pero en la mayoría de los casos tiende a ser invasivo sólo superficialmente, y a tener un curso relativamente benigno. A pesar del acceso a los vasos linfáticos, la enfermedad metastásica es rara, encontrando cifras en la literatura de aproximadamente el 1%. En pacientes inmunosuprimidos (trasplante de órganos, SIDA) existe mayor riesgo de desarrollar este carcinoma y en estos casos son mas agresivos y con mayor potencial metastatizante (1,2,20).

Fig. 8: Carcinoma escamoso de ocnjuntiva con invasión orbitaria
en la zona nasal (A). Las células neoplásicas han roto la membrana
basal e invadido el tejido subconjuntival (B) (HE4x).
Histológicamente la mayoría de los carcinomas son bien diferenciados, con crecimiento exofítico de células epiteliales. En tumores mas avanzados, suele existir inflamación en la sustancia propia, que contiene nidos de células atípicas, que suelen tener núcleos hiperplásicos e hipercromáticos, disqueratosis, perlas corneas (colecciones de células queratinizadas) y mitosis atípicas (1,2,5).
Carcinoma escamoso fusiforme. Son raros y mucho más agresivos, presentándose como lesiones planas con gran agresividad y tendencia a la penetración intraocular, simulando en ocasiones ulceras periféricas corneales, o pterigion. Histológicamente, las células son fusiformes, pleomórficas, con un núcleo hipercromático, a veces difíciles de diferenciar de fibroblastos, pudiendo ser erróneamente diagnosticados como histiocitomas fibrosos o fibrosarcomas. Los estudios immunohistoquimicos positivos para citoqueratinas, confirman la naturaleza epitelial de este tumor (21).
Carcinoma Mucoepidermoide. Son raros y suelen aparecer en ancianos. Son más agresivos que los carcinomas escamosos, y tienden a invadir el globo ocular y la órbita. Suele aparecer en fondo de saco conjuntival, presentando un componente globular amarillento debido al componente de células mucosecretoras. Sus características histológicas son las de una neoplasia epitelial, que contiene una proporción variable de células secretoras de moco, y en ocasiones áreas de diferenciación hacia adenocarcinoma (1).
Tratamiento de los tumores epiteliales
Los objetivos del tratamiento de los tumores de conjuntiva son:
Destruir o extirpar completamente el tumor, mediante cirugía y tratamientos adyuvantes si están indicados y son necesarios (crioterapia, quimioterápicos tópicos, radioterapia).
Realizar un diagnóstico histopatológico preciso, ya que clínicamente es muy difícil distinguir lesiones benignas, precancerosas y malignas. La confirmación diagnóstica permitirá realizar un abordaje terapéutico correcto, así como del pronóstico y seguimiento del paciente.
Minimizar las recurrencias
Para ello se debe realizar una evaluación preoperatoria completa que incluya una aproximación clínica al diagnostico lo mas precisa posible, si la lesión es circunscrita o difusa, uni o bilateral, si se sospecha si es precancerosa o maligna. Se debe evaluar extensión del tumor determinando la existencia de invasión intraocular y/o orbitaria, realizando la palpación de los linfáticos regionales y, cuando se considere indicado estudio de extensión sistémica para detección de metástasis (que son raras) (22).
En general, para los tumores circunscritos, límbicos o de conjuntiva bulbar, la extirpación completa (biopsia escisional) con la mínima manipulación posible y un margen de resección de 3 a 5 mm puede ser tratamiento suficiente. La capa de Bowman debe respetarse ya que su extirpación facilitaría la penetración intraocular de la recurrencia en caso de existir. La crioterapia y el control de los bordes de resección mediante biopsias intraoperatorias han demostrado disminuir las recurrencias en el caso de lesiones precancerosas y malignas (20,22-24).
En las lesiones difusas y extensas, en las que la resección completa es difícil, se debe realizar la extirpación más amplia posible, y que nos permita el diagnóstico histopatológico preciso. Si la resección es muy amplia se pueden realizar injertos de mucosa conjuntival o bucal autóloga. Para el tumor residual, se emplean terapias adyuvantes como, quimioterápicos tópicos (Mitomicina C, 5-Fluorouracilo, interferon) en incluso radioterapia (25,15-17).
Si existe invasión intraocular esta indicada la enucleación; si además hay invasión orbitaria anterior, exenteración anterior, con conservación de los parpados, siempre que la conjuntiva palpebral no esté afectada.
Tumores melanocíticos
Los tumores pigmentados de la conjuntiva representan aproximadamente un 50% de las lesiones tumorales conjuntivales (1,2) y afectan principalmente a pacientes de raza blanca. Estos tumores proceden de los melanocitos que migran desde la cresta neural durante el desarrollo embriológico hasta el epitelio y tejido subconjuntival. Los melanocitos epiteliales son los que se sitúan en la capa basal y son responsables de la mayor parte de las lesiones pigmentadas de la conjuntiva. Comprenden un espectro de alteraciones que abarcan desde lesiones benignas como el nevus de conjuntiva hasta condiciones malignas, potencialmente letales, como el melanoma de conjuntiva. La identificación clínica e histopatológica de estas lesiones es importante para su reconocimiento correcto y tratamiento adecuado (26-27). Lesiones benignas tratadas como malignas, conducen a tratamientos innecesarios, y melanomas o melanosis adquiridas infravaloradas y sin tratar precozmente conducen al acortamiento del pronóstico vital del paciente, evolucionando hacia melanomas que presentan una mortalidad del 25% a los 5 años (26).
Benignos
Nevus congénito/adquirido. Es la lesión melanocítica más común de la conjuntiva. Es congénito y se manifiesta clínicamente durante la infancia como una lesión circunscrita, plana, poco pigmentada, ligeramente elevada en la conjuntiva bulbar interpalpebral (fig. 9A). Suele pigmentarse y aparecer quistes en la segunda década de la vida, momento en que el paciente suele acudir a la consulta (fig. 9B). A partir de esta etapa, los nevus permanecen estables durante la vida adulta, por lo que cualquier cambio en el tamaño, coloración, bordes o apariencia deben hacer sospechar la transformación maligna en melanoma, lo que ocurre en menos del 1% de los nevus (fig. 9C). Su localización más característica es la conjuntiva bulbar, pero también pueden aparecer en el área yuxtalimbal, y en la carúncula. Se mueven libremente sobre la esclera y no se extienden sobre la cornea. Son excepcionales en fondos de saco y conjuntiva tarsal, por lo que cualquier lesión de esta localización debe ser considerada un melanoma o precursor y debe ser biopsiada. Respecto a la pigmentación es muy variable desde marrón claro a chocolate oscuro, y el 30% son no pigmentados (1,26-28).

Fig. 9: Nevus de conjuntiva en la primera década de la vida (A),
en el adulto, con quistes característicos (B) y con cambios de
coloración y bordes: sospecha de malignización (C).
Histológicamente sufren cambios evolutivos, al igual que los de la piel, que se reflejan en los cambios clínicos descritos. En la fase inicial los nidos (tecas) de células névicas se encuentran en la capa basal (nevus epitelial), para más adelante aparecer en la interfase entre el epitelio y el estroma (nevus juncional). A medida que va evolucionando, los nidos de células névicas penetran en el estroma (nevus compuesto), arrastrando células epiteliales que pueden formar pseudoquistes, para terminar localizándose solamente en el tejido subconjuntival (nevus subepitelial o estromal) (1,22,24) (fig. 10).

Fig. 10: Microscopía óptica de nevus conjuntival con tecas
de células juncionales y estromales y quistes de inclusión (HE40x).
Pigmentación Racial. En individuos de raza negra y pigmentación oscura, es frecuente encontrar mayor pigmentación en el epitelio conjuntival. Este pigmento es más prominente en la fisura interpalpebral, es bilateral, se moviliza sobre la superficie del globo ocular, y puede ser marcado en el limbo extendiéndose a la cornea periférica. Microscópicamente se caracteriza por una hiperpigmentacion uniforme de la capa basal del epitelio conjuntival.
Melanocitosis ocular. Es el resultado de la migración incompleta de los melanocitos desde la cresta neural, que no alcanzan la conjuntiva, y pueden aparecer en la úvea, la esclera, epiesclera, nervio óptico, meninges, y la dermis del párpado. Si solo se afecta el globo ocular, se denomina Melanosis Ocular Congénita o Melanosis Oculi. Cuando la pigmentación alcanza la piel del párpado se denomina Melanocitosis oculodérmica o Nevus de Ota.
Clínicamente, es unilateral, y se presenta como una pigmentación difusa gris-azulada epiescleral; los bordes de la pigmentación no se mueven con la conjuntiva (fig. 11A), son espiculados y los melanocitos rodean y dibujan los linfáticos y vasos sanguíneos. La úvea y el iris ipsilateral suelen tener una coloración más oscura produciendo heterocromia tanto de iris como de fondo de ojo (fig. 11B). Histológicamente, los melanocitos están uniformemente pero no intensamente pigmentados, y se sitúan en la esclera y epiesclera más que en la conjuntiva. Aunque esta entidad presenta un riesgo mayor para el desarrollo de glaucoma y de melanoma de órbita y de úvea (1/400) no lo es para melanoma de conjuntiva, ya que no se conoce ningún caso descrito (29,30).

Fig. 11: Melanosis oculi con manchas azules en la esclera
(A) y heterocromía de iris (B).
Melanosis Adquirida secundaria. Es el aumento de pigmentación conjuntival secundario a diferentes causas: enfermedades metabólicas (ej. Enf. Addison), depósitos por drogas instiladas tópicamente, irradiación, inflamación cambios hormonales, trastornos conjuntivales crónicos. No predisponen al desarrollo de melanoma. Histológicamente los melanocitos normales del epitelio, se estimulan, proliferan y producen melanina.
Precancerosos
Nevus con atipia. Como se ha descrito previamente, los nevus son lesiones benignas cuyas células pueden sufrir una transformación maligna a lo largo de su evolución, generalmente en la vida adulta, en mayores de 40 años, asociada a cambios en la morfología clínica que sin tratamiento suele evolucionar a melanoma (fig. 9C) (28).
Melanosis Adquirida primaria (MAP). Es una proliferación melanocítica epitelial unilateral multicéntrica, adquirida que aparece en adultos, que afecta con mayor frecuencia a individuos de raza blanca y piel clara. A lo largo de la literatura ha recibido diferentes nombres como: melanosis precancerosa de Reese, melanosis adquirida benigna o idiopática, hiperplasia melanocítica intraepitelial atípica.
Clínicamente comienza insidiosamente en la edad media de la vida, como una pigmentación sutil, multicéntrica, unilateral extendida por toda la conjuntiva incluyendo fondos de saco y conjuntiva tarsal (fig. 12). Cuando la conjuntiva del borde palpebral se afecta, se extiende a menudo a la epidermis adyacente. Habitualmente es plana, a diferencia de los nevus, y cuando deja de serlo, es un signo de malignización. La coloración es irregular, variando desde la falta de pigmentación hasta marrón oscuro. Su evolución es impredictible. Se puede observar la desaparición de pigmentación en un área y la aparición o aumento en otra. La velocidad de progresión es variable, pero en general es de años (26,31).

Fig. 12: Melanosis Adquirida Primaria. A: Localizada yuxtalímbica.
B y C: Multicéntrica con afectación de fondos de saco,
conjuntiva tarsal, carúncula y borde palpebral.
Histopatológicamente, la Melanosis Adquirida Primaria se clasifica en MAP sin atipia y con atipia (26). El primer grupo se refiere a hiperpigmentación o hiperplasia melanocítica limitada a la capa basal del epitelio. Cualquier tipo de crecimiento de células atípicas diferente a una hiperplasia basilar es considerado una MAP con atipia. Esta diferenciación tiene importancia pronóstica debido a que la MAP con atipia tiene un riesgo de progresión a melanoma aproximadamente de 70-90%, mientras que la MAP sin atipia tiene un riesgo bajo de malignización (20%).

Fig. 13: Melanosis Adquirida Primaria sin atipia. A: Hiperplasia
melanocítica confinada a la capa basal (10x).
B: Hiperpigmentación de la capa basal (HE10x).

Fig. 14: Melanosis Adquirida Primaria con atipia. A: Nidos de
células melanocíticas atípicas que ocupan prácticamente la totalidad
del epitelio con infiltrado inflamatoria subepitelial (HE10x). B: Las células
de estirpe melanocítica marcan positivamente con HMB45 demostrando
que la membrana basal está respetada (HMB4510x).
Melanoma
El melanoma conjuntival es una neoplasia extremadamente rara y potencialmente letal con una incidencia estimada de 0,2 a 0,5 casos por millón de habitantes/año en poblaciones caucásicas. Representa el 1-2% de los tumores malignos oculares. Aunque se ha implicado a la radiación ultravioleta como agente causal, su etiología continúa siendo desconocida. En aproximadamente un 75% de los casos se origina clínicamente de una MAP con atipia (fig. 15A), pero también puede originarse de un nevus previo (20-30%) o aparecer de novo (5-10%) sin ninguna lesión preexistente (fig. 15B) (32-34).
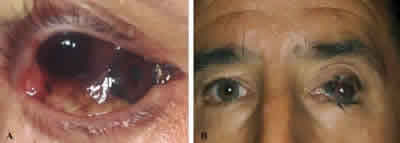
Fig. 15: Melanoma de conjuntiva multicéntrico originado
en una MAP (A), y melanoma de novo avanzado (B).
Afecta pacientes adultos o ancianos de raza blanca y no tiene predilección por el género. La presentación clínica más común es la de una masa elevada, variablemente pigmentada, con signos asociados de MAP o historia de nevus, que se localiza en limbo, conjuntiva bulbar, fornix o conjuntiva palpebral. Puede extenderse localmente a la órbita y el globo ocular y sistémicamente a través de los linfáticos, por lo que previamente al tratamiento es importante determinar el grado de extensión tumoral, realizando un completo examen ocular y orbitario, palpación de ganglios de cuello y estudio de extensión sistémica.
Histopatológicamente el melanoma de conjuntiva se compone de células melanocíticas atípicas de morfología muy diversa, que se encuentran invadiendo la sustancia propia conjuntival (fig. 16). La morfología es muy variable presentando desde células pleomórficas, grandes con nucleolos prominentes hasta pequeñas células poliédricas o fusiformes sin pigmento identificable. La inmunohistoquímica para proteína S-100 y HMB-45 puede ayudar a diagnosticar casos indiferenciados y amelanóticos (1,29-34).

Fig. 16: Melanoma de conjuntiva. A: Proliferación celular en
el tejido subconjuntival con pigmentación irregular (HE4x).
B: A mayor aumento (HE40x) se observan células atípicas,
pleomórficas, de estirpe epitelioide y pigmentación variable con numerosas mitosis.
A pesar de un tratamiento adecuado, aproximadamente la mitad de los pacientes presentan recurrencias locales y un tercio de los casos metástasis a distancia a los 10 años (32,33), por lo que es importante un seguimiento continuado de estos pacientes. La localización mas frecuente de metástasis iniciales son los nódulos linfáticos regionales (preauriculares, submandibulares y cervicales), aunque también ocurre diseminación a distancia por vía hematógena. Una vez que se presenta la enfermedad diseminada, no existe tratamiento eficaz y los pacientes tienen un tiempo de supervivencia relativamente corto. Los factores pronósticos mas importantes son el espesor del tumor, la localización palpebral o en carúncula, la invasión linfática, índice de proliferación elevado, y el patrón histológico pagetoide y las recurrencias (1,26,32-34). Actualmente, se están investigando diferentes aspectos, como la importancia de la biopsia del ganglio linfático centinela en la detección precoz de metástasis regionales (35,37), el reconocimiento de factores de riesgo para el desarrollo tumoral y la evaluación de marcadores pronóstico de morbi-mortalidad (5,34,37,38).
Tratamiento de los tumores melanocíticos
Los objetivos generales del tratamiento de los tumores melanocíticos son similares a los descritos para los epiteliales: extirpación o destrucción completa del tumor, diagnóstico histopatológico preciso y minimizar las recurrencias, objetivo especialmente importante en los tumores pigmentados ya que empeoran el pronóstico del paciente.
Las lesiones benignas, en general no requieren más que un diagnóstico clínico correcto y la observación periódica. La mayoría de los nevus por debajo de los 40 años no requieren tratamiento excepto por motivos estéticos, o procesos inflamatorios de repetición, ya que el melanoma es extremadamente raro en jóvenes. Incluso en personas mayores, solo en el caso de crecimiento o cambios en el color y morfología debe realizarse la extirpación de la lesión para descartar transformación maligna.
El manejo de la MAP depende la extensión de la lesión. Si es pequeña, ocupando menos de un cuarto de conjuntiva, se debe plantear su extirpación completa. Si la lesión es muy extensa, se deben extirpar las áreas engrosadas (sospechosas de melanoma) y realizar biopsias en mapa de las zonas de MAP no extirpadas para determinar su riesgo de progresión a melanoma de conjuntiva. Incluso, deben realizarse biopsias en zonas aparentemente no involucradas debido a la presencia de MAP sine pigmento. Si la extirpación completa de las zonas con atipia no es posible o aparecen recurrencias, se deben utilizar terapias adyuvantes como la crioterapia intraoperatoria o tratamiento quimioterápico tópico post-quirúrgico de los que el más probado es la Mitomicina C (fig. 17) (32,39-41).

Fig. 17: Melanoma multicéntrico en MAP (A). Postratamiento
con extirpación nódulos engrosados, biopsia en mapa y tratamiento
con Mitomicina tópica de la pigmentación residual (B).
En el melanoma de novo o procedente de un nevus, el tratamiento primario del tumor es la extirpación quirúrgica completa, con 3-5 mm de márgenes libres con o sin crioterapia intraoperatoria adyuvante. Se debe evitar realizar biopsias incisionales. Cualquiera que sea el origen del melanoma, se debe de tratar de erradicar combinando las terapias necesarias en el primer abordaje para prevenir las recurrencias, ya que estas empeoran el pronóstico vital del paciente.
En el seguimiento, los pacientes deben ser cuidadosamente revisados en la consulta, explorando la superficie entera de la conjuntiva exhaustivamente y palpando los ganglios preauriculares y cervicales. Si se identifican invasión ipsilateral preauricular, submandibular y/o cervical, se recomienda la realización de una limpieza ganglionar regional no radical.
Si se objetiva invasión orbitaria y/o intraocular, el tratamiento primario generalmente es la exenteración. La enucleación no se suele realizar, ya que deja la conjuntiva, que es el lugar del origen del tumor y puede ser fuente de nuevas recurrencias.
Tumores de tejidos blandos, glándulas y anejos
Los tejidos no epiteliales de la conjuntiva incluyen estructuras vasculares y linfáticas, nervios periféricos, elementos conectivos, glándulas y anejos cutáneos en la carúncula, que pueden dar lugar a cualquier tumor de tejidos blandos que pueda surgir en cualquier parte de la economía, con las mismas características histopatológicas, aunque la mayoría de ellos son muy raros. Sarcomas, fibromas, neurofibromas, schwanomas, neurotekiomas. y otros tumores de partes blandas pueden aparecer en la conjuntiva pero y con mucha más frecuencia, la conjuntiva es un lugar de afectación secundario de la afectación orbitaria (1,2,42,43). Se describen a continuación los más frecuentes y/o relevantes.
Benignos
Carúncula: Oncocitoma o cistadenoma eosinofílico. La mayoría de los tumores que se originan en las estructuras anejas de los parpados, las cejas y la orbita, pueden surgir en la carúncula (ej. adenoma pleomórfico, oncocitoma, carcinoma de glándula sudorípara, carcinoma sebáceo). Este hecho no es sorprendente, ya que la carúncula contiene glándulas lagrimales accesorias, glándulas sebáceas y folículos pilosos. El tumor maligno mas frecuente de la carúncula es el carcinoma sebáceo y el benigno, el oncocitoma que también aparece en la glándula y saco lagrimal. Se presenta como una masa sonrosada elevada de crecimiento lento en personas mayores (1,42).
Microscópicamente los oncocitomas tienen un patrón variable, y las células pueden disponerse en planos, cordones o nidos o pueden formar estructuras quísticas o glandulares. Las células que lo componen típicamente tienen el citoplasma amplio eosinofílico, lleno de material que en microscopia electrónica son mitocondrias. Esta transformación oncocítica no es específica, y ocurre en otras glándulas y mucosas del cuerpo, y puede representar un cambio de envejecimiento.
Granuloma piogénico. Clínicamente se presenta en como una lesión pedunculada y aspecto papilomatoso de color sonrosado que se desarrolla después de un trauma quirúrgico o accidental o un proceso inflamatorio local (chalazión o reacción a cuerpo extraño). El diagnóstico diferencial debe realizarse principalmente con un papiloma pediculado, con el que generalmente le diferencia la velocidad de progresión y el antecedente de traumatismo o chalacion. Histopatológicamente consiste en tejido de granulación compuesto por un estroma laxo conteniendo numerosos capilares que se distribuyen de forma radial, y una mezcla de células inflamatorias agudas y crónicas (fig. 18) (1). Su tratamiento consiste en la biopsia escisional (4).

Fig. 18: Granuloma Piogénico tras trauma quirúrgico. A: Formación exofítica,
sonrosada, ricamente vascularizada. B: Tejido de granulación con numerosos
capilares dispuestos radialmente (HE4x).
Hemangioma. Son tumores que consisten en una proliferación vascular benigna que pueden ser localizados en la propia conjuntiva o formar parte de hemangiomas que afectan a otras estructuras. La conjuntiva es un lugar poco habitual pero se suele ver afectada en los hemangiomas capilares de órbita y párpados que aparecen precozmente pero regresan espontáneamente en los primeros años de vida, y también en los síndromes de Sturge-Weber que suelen presentar hemangiomas difusos conjuntivales ipsilaterales (1).
Linfangiectasia/Linfangioma. Consiste en la dilatación de los vasos linfáticos de la conjuntiva, que puede aparecer en la conjuntiva bulbar o tarsal. Cuando aparecen como una masa irregular, rojiza compuesta de numerosos linfáticos dilatados que pueden contener sangre y progresan lentamente, y se exacerban con procesos catarrales, son linfangiomas, que suelen formar parte de una lesión que afecta también a la órbita y los párpados (fig. 19) (1).

Fig. 19: Linfangioma de conjuntiva. A: Dilatación de vasos linfáticos
extensa que infiltra conjuntiva bulbar, fondo de saco y carúncula, que
confluyen y se hacen prominentes. B: Linfáticos dilatados y tortuosos
parcialmente llenos de material proteináceo (HE10x).
Malignos
Carúncula: Carcinoma de células sebáceas. Es raro, pero según el Instituto de las Fuerzas Armadas de Washington es el tumor maligno mas frecuente de la carúncula (1). Se origina en las glándulas sebáceas, y su diseminación pagetoide sustituyendo al epitelio conjuntival puede simular una conjuntivitis unilateral. La conjuntiva también es frecuentemente invadida por esta diseminación superficial de un carcinoma sebáceo originado en los parpados.
Sarcoma de Kaposi. Ha sido una enfermedad rara hasta hace pocos años, que ocurría en individuos mayores, asociada a judíos-mediterráneos. Actualmente se observa en individuos jóvenes afectados por SIDA y puede ser una de las manifestaciones precoces de esta enfermedad. Clínicamente consiste en la aparición de nódulos o manchas rojo-azuladas en la piel de las piernas en los pacientes con la forma clásica de la enfermedad, pero en aquellos con Síndrome de Inmunodeficiencia Adquirida (SIDA) las lesiones aparecen en la parte superior del cuerpo, en la cara, los párpados, y la afectación conjuntival puede ser el primer signo clínico de SIDA. Aparecen como nódulos subepiteliales en fornix o conjuntiva palpebral. Histológicamente están formados por células fusiformes de núcleo oval y numerosos capilares (fig. 20) (1).

Fig. 20: Sarcoma de Kaposi. A: Nódulo conjuntival subepitelial
rojizo de límites poco definidos. B: Celularidad fusiforme con
numerosas hendiduras capilares (HE40x).
Tumores linfoides
Los linfomas del ojo y anejos oculares suponen entre del 2 al 10% de los linfomas extranodales (son raros, y comprenden un espectro que abarca desde la hiperplasia reactiva linfoide al linfoma maligno basados siempre en características microscópicas. Los linfomas de conjuntiva suponen entre el 20 y el 30% de los tumores linfoides del ojo y anejos, y tan solo un 20-30% de los casos se encuentra asociado a enfermedad sistémica (que puede aparecer años después), siendo bilaterales en casi el 40% de los casos (44,45).
La mayoría de los linfomas de los anejos oculares son de células B tipo no Hodking de bajo grado, y la mayor parte de la conjuntiva tipo MALT (Linfoma B de bajo grado del tejido linfoide asociado a mucosas) situándose en el extremo mas favorable de los linfomas malignos, y en muchas ocasiones, en un terreno difícil para el diagnóstico diferencial en el que no hay una clara línea de corte entre el linfoma maligno de bajo grado y la hiperplasia reactiva linfoide.
Clínicamente tanto las lesiones benignas como las malignas tienden a tener una coloración salmón, y suelen ser lesiones subconjuntivales, planas con superficie lisa y de consistencia blanda, que se localizan cerca de los fórnices. Clínicamente es imposible diferenciar entre lesiones benignas (hiperplasia reactiva linfoide) y malignas, por lo que es imprescindible la realización de una biopsia, y si se confirma el diagnóstico de lesión maligna, la realización de una exploración sistémica del paciente para excluir la presencia de enfermedad sistémica (fig. 21).

Fig. 21: Linfoma localizado unilateral de conjuntiva tipo MALT.
A: Masa asalmonada subconjuntival infiltrativa que afecta fondo de saco,
conjuntiva tarsal. B y C: Microscópicamente se observa un denso infiltrado
subepitelial compuesto por células pequeñas de estirpe linfocítica, en
su mayoría linfocitos B (HE4x y 40x aumentos respectivamente).
El tratamiento adecuado si hay linfoma sistémico es la quimioterapia sistémica, a la que la afectación conjuntival responderá de la misma forma que las zonas afectadas del resto del organismo. Si no hay afectación sistémica y la lesión está localizada en la conjuntiva se puede realizar extirpación quirúrgica, radioterapia externa si la lesión es extensa o se sospecha afectación orbitaria (20-40Gy), inyecciones locales de interferon e incluso la observación (44-47).
Sumario
En resumen una gran variedad de tumores puede aparecer en la conjuntiva bulbar, tarsal, fondos de saco, pliegue semilunar o carúncula afectando el epitelio o el tejido subepitelial. La mayoría son fáciles de detectar en la exploración oftalmológica rutinaria y su carácter benigno permite un tratamiento conservador o mínimamente invasivo. Sin embargo los más agresivos de carácter premaligno o francamente malignos tales como carcinomas, melanomas o linfomas, que ponen en peligro la función visual y/o la vida del paciente, no son excepcionales y por ello se debe mantener una actitud vigilante para sospechar su presencia y detectarlos de forma precoz. En estos casos su existencia debe ser confirmada mediante una biopsia y la cirugía combinada con distintos tratamientos adyuvantes (quimioterapia, crioterapia u otras) persigue el control local de la enfermedad, preservando en lo posible la integridad anatómica y funcional de la superficie ocular. Las nuevas técnicas de imagen y algunas métodos tales como el estudio del ganglio centinela tienen importancia para detectar la extensión local o a distancia de la enfermedad. A pesar de todos los avances diagnósticos y terapéuticos el fundamento de un correcto abordaje en el tratamiento de los tumores conjuntivales sigue dependiendo de un buen estudio y diagnóstico histopatológico para lo que es primordial la correlación clínico-patológica que exige una buena comunicación entre el oftalmólogo y el patólogo.
Bibliografía
1. Font RL, Croxatto O, Rao N. Tumors of the eye and ocular adnexa. Washington: American Registry of Pathology & Armed Forces Institute of Pathology. 2006; 1-40. [ Links ]
2. Shields CL, Shields JA. Tumors of the conjunctiva and cornea. Surv Ophthalmol 2004; 49: 3-24. [ Links ]
3. Portero-Benito A, Sánchez-Mannarelli F, Carreño-Salas E, Muñoz MF, Saornil MA. Spectrum of conjunctival tumors in an ocular oncology unit: review of 314 cases. Acta Ophthalmol Scand 2008; S243: 667. [ Links ]
4. Sánchez-Mannarelli F, Portero-Benito A, Carreño-Salas E, Muñoz MF, Saornil MA. Frequency of melanocytic conjunctival lesios in an ocular oncology unit. Acta Ophthalmol Scand 2008; S243: 4461. [ Links ]
5. Rodrigues MM, Hidayat AA. Conjunctival and corneal pathology. In: Albert DM, Jakobiec FJ. Principles and practice of ophthalmology. Philadelphia: WB Saunders Co. 2000; Vol. 4: 3609-3633. [ Links ]
6. Cibis GW. Basic and Clinical Science Course. Section 2. 2005-2006, Fundamentals and principles of ophthalmology. San Francisco: American Academy of Ophthalmology; 2005; 528. [ Links ]
7. Knop N, Knop E. Conjunctiva-associated lymphoid tissue in the human eye. Invest Ophthalmol Vis Sci 2000; 41: 1270-1279. [ Links ]
8. Grossniklaus HE. Basic and Clinical Science Course. Section 4. 2005-2006, Ophthalmic pathology and intraocular tumors. San Francisco: American Academy of Ophthalmology; 2005; 332. [ Links ]
9. Mauriello JA Jr, Napolitano J, McLean I. Actinic keratosis and dysplasia of the conjunctiva: a clinicopathological study of 45 cases. Can J Ophthalmol 1995; 30: 312-316. [ Links ]
10. Napora C, Cohen EJ, Genvert GI, Presson AC, Arentsen JJ, Eagle RC, et al. Factors associated with conjunctival intraepithelial neoplasia: a case control study. Ophthalmic Surg 1990; 21: 27-30. [ Links ]
11. Scott IU, Karp CL, Nuovo GJ. Human papillomavirus 16 and 18 expression in conjunctival intraepithelial neoplasia. Ophthalmology 2002; 109: 542-547. [ Links ]
12. Wilson MW, Hungerford JL, George SM, Madreperla SA. Topical mitomicyn C for the treatment of conjunctival and corneal epithelial dysplasia and neoplasia. Am J Ophthalmol 1997; 124: 303-311. [ Links ]
13. Wilson MW, Czechonska G, Finger PT, Rausen A, Hooper ME, Haik BG. Chemotherapy for eye cancer. Surv Ophthalmol 2001; 45: 416-444. [ Links ]
14. Yeatts RP, Engelbrecht NE, Curry CD, Ford JG, Walter KA. 5-Fluorouracil for the treatment of intraepithelial neoplasia of the conjunctiva and cornea. Ophthalmology 2000; 107: 2190-2195. [ Links ]
15. Karp CL, Moore JK, Rosa RH Jr. Treatment of conjunctival and corneal intraepithelial neoplasia with topical interferon alpha-2b. Ophthalmology 2001; 108: 1093-1098. [ Links ]
16. Sturges A, Butt AL, Lai JE, Chodosh J. Topical interferon or surgical excision for the management of primary ocular surface squamous neoplasia. Ophthalmology 2008; 115: 1297-1302. [ Links ]
17. Toledano Fernández N, García Sáenz S, Díaz Valle D, Arteaga Sánchez A, Segura Bedmar M, Lorenzo Giménez S, et al. Empleo de interferon Alfa-2B para el tratamiento de carcinomas conjuntivales intraepiteliales en casos seleccionados. Arch Soc Esp Oftalmol 2003; 78: 265-271. [ Links ]
18. Arteaga-Sánchez A, Toledano-Fernández N, Díaz-Valle D, Fernández-Aceñero MJ, Hijós-Gastón M. Escleroqueratitis y carcinoma escamoso conjuntival invasivo. Arch Soc Esp Oftalmol 2007; 82: 237-240. [ Links ]
19. López García JS, Elosúa De Juan I, González Morales ML, de Pablo Martín C, Alvarez Lledo J, Martínez Garchitorena J. Carcinoma de células escamosas conjuntival con invasión orbitaria. Arch Soc Esp Oftalmol 2000; 75: 637-641. [ Links ]
20. Tunc M, Char DH, Crawford B, Miller T. Intraepithelial and invasive squamous cell carcinoma of the conjunctiva: analysis of 60 cases. Br J Ophthalmol 1999; 83: 98-103. [ Links ]
21. Seregard S, Kock E. Squamous spindle cell carcinoma of the conjunctiva. Fatal outcome of a plerygium-like lesion. Acta Ophthalmol Scand 1995; 73: 464-466. [ Links ]
22. Shields JA, Shields CL, De Potter P. Surgical management of conjunctival tumors. The 1994 Lynn B. McMahan Lecture. Arch Ophthalmol 1997; 115: 808-815. [ Links ]
23. Sudesh S, Rapuano CJ, Cohen EJ, Eagle RC Jr, Laibson PR. Surgical management of ocular surface squamous neoplasms. The experience from a cornea center. Cornea 2000; 19: 278-283. [ Links ]
24. Damato B. Ocular Tumors. Diagnosis and treatment. Oxford: Butterworth Heineman; 2000. [ Links ]
25. Majmudar PA, Epstein RJ. Antimetabolites in ocular surface neoplasia. Curr Opin Ophthalmol 1998; 9: 35-39. [ Links ]
26. Jakobiec FA, Folberg R, Iwamoto T. Clinicopathologic characteristics of premalignant and malignant melanocytic lesions of the conjunctiva. Ophthalmology 1989; 96 :147-166. [ Links ]
27. Shields CL, Demirci H, Karatza E, Shields JA. Clinical survey of 1643 melanocytic and nonmelanocytic conjunctival tumors. Ophthalmology 2004; 111: 1747-1754. [ Links ]
28. Folberg R, Jakobiec FA, Bernardino VB, Iwamoto T. Benign conjunctival melanocytic lesions. Clinicopathologic features. Ophthalmology 1989; 96: 436-461. [ Links ]
29. Singh AD, De Potter P, Fijal BA, Shields JA, Elston RC. Lifetime prevalence of uveal melanoma in white patients with oculo (dermal) melanocytosis. Ophthalmology 1998; 105: 195-198. [ Links ]
30. López-Caballero C, Saornil-Alvarez MA, Blanco-Mateos G, Frutos-Baraja JM, López-Lara F, González-Sansegundo C. Melanoma de coroides en melanosis óculi. Arch Soc Esp Oftalmol 2003; 78: 99-102. [ Links ]
31. Folberg R, McLean IW, Zimmerman LE. Conjunctival melanosis and melanoma. Ophthalmology 1984; 91: 673-678. [ Links ]
32. Seregard S. Conjunctival melanoma. Surv Ophthalmol 1998; 42: 321-350. [ Links ]
33. Shields CL. Conjunctival melanoma: risk factors for recurrence, exenteration, metastasis, and death in 150 consecutive patients. Trans Am Ophthalmol Soc 2000; 98: 471-492. [ Links ]
34. Paridaens AD, Minassian DC, McCartney AC, Hungerford JL. Prognostic factors in primary malignant melanoma of the conjunctiva: a clinicopathological study of 256 cases. Br J Ophthalmol 1994; 78: 252-259. [ Links ]
35. Nijhawan N, Ross MI, Diba R, Ahmadi MA, Esmaeli B. Experience with sentinel lymph node biopsy for eyelid and conjunctival malignancies at a cancer center. Ophthal Plast Reconstr Surg 2004; 20: 291-295. [ Links ]
36. Baroody M, Holds JB, Kokoska MS, Boyd J. Conjunctival melanoma metastasis diagnosed by sentinel lymph node biopsy. Am J Ophthalmol 2004; 137: 1147-1149. [ Links ]
37. Tuomaala S, Kivelä T. Metastatic pattern and survival in disseminated conjunctival melanoma: implications for sentinel lymph node biopsy. Ophthalmology 2004; 111: 816-821. [ Links ]
38. Tuomaala S, Eskelin S, Tarkkanen A, Kivelä T. Population-based assessment of clinical characteristics predicting outcome of conjunctival melanoma in whites. Invest Ophthalmol Vis Sci 2002; 43: 3399-3408. [ Links ]
39. Becerra EM, Blanco G, Saornil MA, del C Méndez M, Bianciotto CG. Hughes technique, amniotic membrane allograft, and topical chemotherapy in conjunctival melanoma with eyelid involvement. Ophthal Plast Reconstr Surg 2005; 21: 238-240. [ Links ]
40. Demirci H, McCormick SA, Finger PT. Topical mitomycin chemotheraphy for conjunctival malignant melanoma and primary acquired melanosis with atypia: clinical experience with histopathologic observations. Arch Ophthalmol 2000; 118: 885-891. [ Links ]
41. Frucht-Pery J, Peer J. Use of mitomicin C in the treatment of conjunctival primary adquired melanosis with atipia. Arch Ophthalmol 1996; 114: 1261-1264. [ Links ]
42. Toledano Fernández N, García Sáenz S, Arteaga Sánchez A, Jareño Dorrego ME, Román Guindo JM. Onocitoma de la carúncula. Caso clínico. Arch Soc Esp Oftalmol 2000; 75: 709-712. [ Links ]
43. Toledano Fernández N, García Sáenz S, Varona Crespo C, Marzabeitia Arambarri F, Arteaga Sánchez A, Díaz Valle D. Neurotekeoma cellular de la conjuntiva bulbar. Caso clínico. Arch Soc Esp Oftalmol 2000; 75: 43-46. [ Links ]
44. Shields CL, Shields JA, Carvalho C, Rundle P, Smith AF. Conjunctival lymphoid tumors: clinical analysis of 117 cases and relationship to systemic lymphoma. Ophthalmology 2001; 108: 979-984. [ Links ]
45. Rubin PA, Kent CJ, Jakobiec FA. Orbital and ocular adnexal lymphoid tumors. In: Albert DM, Jakobiec FA. Principles and practice of ophthalmology. Philadelphia: WB Saunders. 2000; 3182-3197. [ Links ]
46. Blasi MA, Gherlinzoni F, Calvisi G, Sasso P, Tani M, Cellini M, Balestrazzi E. Local chemotheraphy with interferon-alfa for conjunctival mucosa-associated lymphoid tissue lymphoma: a preliminary report. Ophthalmology 2001; 108: 559-562. [ Links ]
47. Acero Peña A, Domingo Gordo B, Arrevola Velasco L, Gómez García J, Martínez Montero JC. Linfoma de la zona marginal de la conjuntiva: carasterísticas clínico-patológicas. Arch Soc Esp Oftalmol 2000; 75: 477-480. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
M. A. Saornil
Hospital Clínico Universitario
Ramón y Cajal, 3
47005 Valladolid
España
E-mail: saornil@ono.com
Recibido: 26/6/07.
Aceptado: 7/1/09.

