










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCCIÓN
Según el panorama descrito por la Estrategia de Seguridad Nacional 2017, en los últimos años, el número de enfermedades emergentes identificadas y de situaciones de riesgo asociadas a ellas ha aumentado. Se han identificado al menos seis alertas sanitarias globales, todas ellas con un importante impacto a nivel nacional: el Síndrome Respiratorio Agudo Grave, la gripe por virus A/H5N1, la pandemia de gripe por virus A/H1N1, la nueva diseminación internacional del poliovirus salvaje, la enfermedad por virus Ébola en África del Oeste y la infección por virus Zika1.
Por otro lado, Europa y el mundo en su conjunto, viven una de las mayores crisis migratorias registradas desde la Segunda Guerra Mundial. En este drama humano se ha incrementado exponencialmente la migración por motivos económicos o relacionados con factores medioambientales. España, por su posición geoestratégica, está especialmente expuesta a este desafío. Prevenir, controlar y ordenar los flujos migratorios irregulares en las fronteras, así como garantizar una adecuada acogida e integración de los inmigrantes, ayuda al control de posibles enfermedades infecciosas de los países de origen1.
España, un país que recibe más de 75 millones de turistas al año, con puertos y aeropuertos que se cuentan entre los de mayor tráfico del mundo, un clima que favorece, cada vez más, la extensión de vectores de enfermedades, con una población envejecida y una situación geopolítica polarizada, no está exenta de amenazas y desafíos asociados a enfermedades infecciosas tanto naturales como intencionadas. En este escenario, resulta primordial implementar acciones para minimizar riesgos, siendo preciso, además de reducir la vulnerabilidad de la población, desarrollar planes de preparación y respuestas ante amenazas y desafíos sanitarios, tanto genéricos como específicos1.
Entre las diversas amenazas conviene destacar las relacionadas con la aparición y diseminación de agentes infecciosos entre la población. La capacidad de respuesta frente a los agentes infecciosos víricos resulta más limitada que la disponible frente a otros agentes patógenos (como bacterias, hongos, protozoos o incluso helmintos); además, los productos antivirales presentan una superior interacción con las células huésped, lo que se traduce en una mayor toxicidad.
El uso de toxinas y posibles agentes vivos para mejorar la capacidad de armas convencionales data de tiempo atrás. Las armas biológicas se caracterizan por su amplio potencial actuando sobre un gran número de personas en áreas amplias; de manera general, los requerimientos logísticos de este tipo de armas son mínimos y su detectabilidad es reducida2. Los agentes infecciosos que pueden ser potencialmente empleados en armas biológicas son variados, entre ellos cabe la posibilidad de utilizar agentes patógenos virales causantes de enfermedades emergentes.
En la Tabla 1 se muestra la gran variabilidad de agentes virales infecciosos, teniendo en consideración diversas características de los mismos relacionadas con su capacidad de generar riesgos relacionados con la salud en la población2.
Tabla 1 Agentes virales infecciosos.
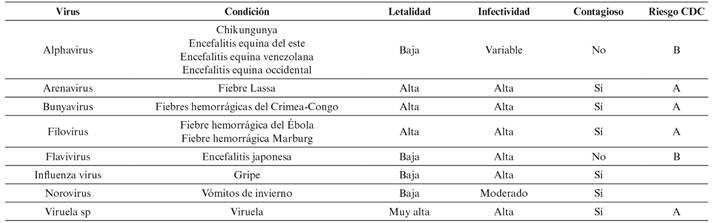
Riesgo CDC (Clasificación de los centros de control de enfermedades): A: alta letalidad, alta infectividad y se contagia persona-persona; B: letalidad moderada, infectividad moderada, interés en salud pública, posible contagio persona-persona; C: amenaza emergente, potencial para usar en liberación deliberada
Entre las enfermedades infecciosas causadas por agentes víricos, resulta destacable el conjunto que se agrupa bajo la denominación de fiebres hemorrágicas víricas, que abarcan desde enfermedades autolimitadas, no transmisibles y leves (dengue) hasta enfermedades altamente contagiosas (Ébola, Lassa, Marburg y fiebre de Crimea-Congo). Todas son zoonosis y tienen como reservorios a animales, con insectos como vectores o exposición a excreciones animales como mecanismos de transmisión. La transmisión de persona a persona puede producirse por exposición a la sangre y fluidos corporales infectados, la aerosolización durante intervención médica, incluyendo hematemesis y hemoptisis, la inoculación o la vía gastrointestinal2.
La clínica observada tras la infección se manifiesta con síntomas difusos como malestar, decaimiento, aumento de la permeabilidad vascular que puede dar lugar a manifestaciones hemorrágicas como hematomas y sangrados, fiebres muy altas (con una duración mayor a 7 días)3. Dependiendo del virus infectante, la clínica se desarrolla según se indica en la Tabla 2 2.
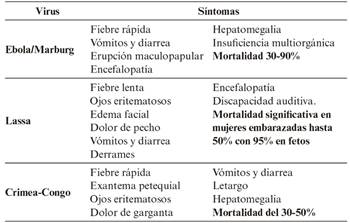
Resulta de interés disponer de medidas que permitan hacer frente a una contingencia provocada por la diseminación de un agente viral altamente infeccioso.
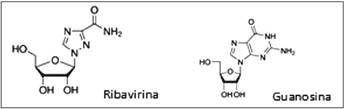
La ribavirina (1-β-D-ribofuranosil-1H-1,2,4-triazol-3-carboxamida) es un nucleósido sintético de la guanosina (Figura 1). Esta molécula se ha planteado como posible contramedida frente a las fiebres hemorrágicas descritas anteriormente2-7. Se trata de un antivírico de amplio espectro que in vitro se muestra activo frente a DNA y RNA vírico, mediante una inhibición de su replicación.
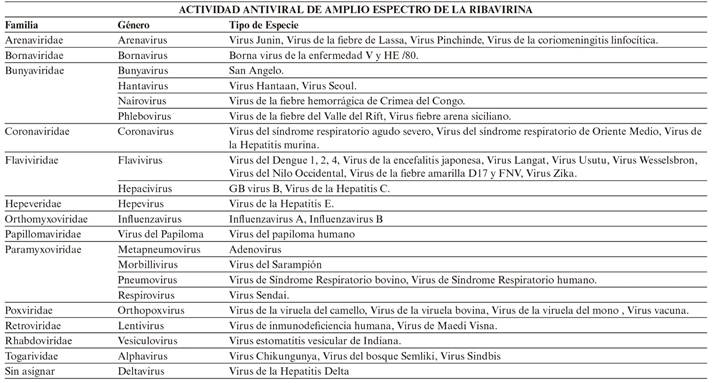
Son sensibles a la acción de la ribavirina el virus respiratorio sincitial (VRS), influenza A y B, parainfluenza, adenovirus y algunos togavirus (rubéola), bunyavirus (Hantavirus) y arenavirus (fiebre de Lassa). También tiene acción in vitro sobre los virus de la hepatitis B y C, virus del herpes simple (VHS) e incluso virus de la inmunodeficiencia humana (VIH)8. El amplio espectro de actividad antiviral de la ribavirina puede ser potencialmente atribuido a sus múltiples mecanismos de acción, pudiendo actuar como análogo de una base purínica en distintos procesos celulares y virales, provocando la activación de múltiples mecanismos simultáneamente9. Sin embargo, sigue siendo una incógnita el mecanismo de acción concreto de la ribavirina, y continúa siendo objeto de múltiples estudios, como los llevados a cabo para probar su eficacia en pacientes infectados con el virus VIH4,8,10.
En la tabla 3 se indican los agentes víricos frente a los cuales la ribavirina es tratamiento de elección11.
La ribavirina se encuentra disponible en diferentes formas de administración oral (comprimidos y cápsulas), aerosol y pomada9,12,13. Desde 1980, en EEUU se empezaron a realizar usos esporádicos de ribavirina intravenosa para pacientes con enfermedades infecciosas graves que podían conducir a la muerte12.
Ante la necesidad de un tratamiento de urgencia frente a los agentes víricos anteriormente citados, la administración intravenosa se configura como la vía más adecuada de administración, no disponiéndose en la actualidad de una forma farmacéutica inyectable comercializada en España. El desarrollo y obtención de una solución inyectable de ribavirina daría lugar a la cobertura de una necesidad terapéutica.
La administración de ribavirina se lleva a cabo de acuerdo a la posología indicada en la Tabla 4, según el agente infeccioso.
Tabla 4 Posología Ribavirina.
| Agente infeccioso | Adultos | Niños |
|---|---|---|
| Fiebre de Lassa | 2 g IV inicial + 1 g IV/6 h (4 días). | |
| Fiebre hemorrágica con síndrome renal secundario | 33 mg/kg IV inicial, seguido 16 mg/kg IV cada 6 h durante 4 días y 8 mg/Kg IV cada 8 horas durante 3 días21. | |
| Crimea Congo | 30 mg/kg IV inicial ,seguida de 16 mg/kg cada 6 horas durante 4 días y, posteriormente y 8 mg/kg cada 8 horas durante 6 días22. | |
| Infección grave de Virus Respiratorio sincitial | Vía inhalatoria 20 mg/ml, 300 ml durante 12-18 h, con un generador de aerosol de partículas pequeñas, durante 3-7 días23. | |
| Hepatitis A | Oral:400 mg 3 veces al día durante 10 días24. | |
| Hepatitis B o C agudas | Oral:400 mg 3 veces al día, durante 20 días24. | |
| Hepatitis C crónica (en combinación con interferón alfa-2b) | Oral: 400 mg 3 veces al día, durante 48 semanas, dependiendo de la respuesta en los parámetros séricos; reducir paulatinamente la dosis hasta suspenderlo24. |
De forma adicional y complementaria siempre se debe reforzar el tratamiento con sueroterapia, analgesia, antibióticos y transfusiones, dependiendo de cada caso.
Como restricciones al empleo de ribavirina, se han descrito por vía inhalatoria posible irritación conjuntival, erupción cutánea y deterioro de las pruebas de función respiratoria como efectos adversos8. Con dosis orales altas o por vía intravenosa se ha observado anemia normocítica normocroma, generalmente reversible, que se sigue de reticulocitosis al suspender el tratamiento8,14. Con tratamientos prolongados se han descrito alteraciones gastrointestinales y neurológicas (cefalea, insomnio y somnolencia) y la ausencia de eliminación en pacientes con insuficiencia renal8,15. Tiene capacidad teratógena en animales de experimentación. Las interacciones medicamentosas son escasas; se ha propuesto cierto antagonismo con la azitromicina8.
OBJETIVO
El objetivo general de este trabajo consiste en el desarrollo de una solución inyectable de ribavirina de 100 mg/ml (10%) en ampollas en el Centro Militar de Farmacia de la Defensa (CEMILFARDEF), para su uso como tratamiento de urgencia frente agentes víricos altamente infecciosos, siendo una terapia de interés estratégico en materia de defensa, seguridad y salud pública.
Los objetivos específicos de este trabajo son:
El desarrollo galénico de una solución acuosa estéril de ribavirina susceptible de administración parenteral.
Estudiar su estabilidad química en ampollas de 5 ml en condiciones «a tiempo real» (largo plazo) durante 12 meses y «aceleradas» (corto plazo) durante 6 meses según Guide International Conference of Harmonisation (ICH) Q1A(R2) «Stability testing of new drug substances and products».
MATERIAL Y MÉTODO
Método
La ribavirina (materia prima) tiene apariencia de polvo cristalino blanco o casi blanco, muy soluble en agua y ligeramente soluble en alcohol 96% y cloruro de metileno. Su pH en solución se encuentra entre 4,0-6,516.
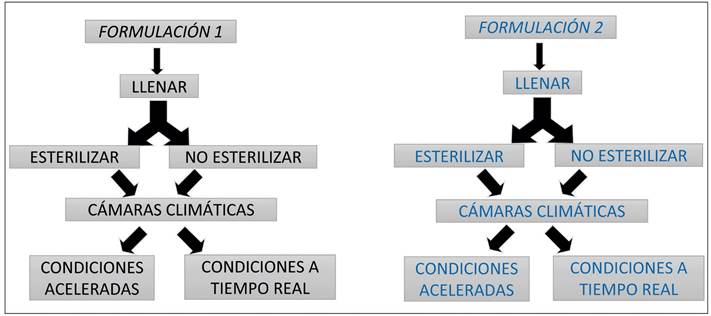
Se establece un desarrollo galénico de dos formulaciones distintas (Figura 2).
1. Preparación
Se plantean dos formulaciones de ribavirina, buscando encontrar la de mayor estabilidad química:
-
Formulación 1 (F1): Ribavirina en agua.
Se prepara una solución intermedia de 1.250 ml, disolviendo 125 gramos del principio activo en agua para inyectables, obteniendo un pH = 3,88.
-
Formulación 2 (F2): Ribavirina con tampón dihidrogenofosfato de sodio anhidro e hidrogenofosfato de sodio anhidro en agua para inyectables.
Se prepara una solución intermedia de 1.250 ml, disolviendo 125 gramos de principio activo con 8 gr de dihidrogenofosfato de sodio anhidro y 2,36 gr de hidrogenofosfato de sodio anhidro, obteniendo un pH = 6,35.
El pH es un factor importante a tener en cuenta en la formulación de soluciones acuosas de administración parenteral, puesto que puede condicionar no solo la tolerancia de la preparación por el organismo, sino también su estabilidad y la actividad del principio activo. El pH de la sangre, la linfa y el líquido cefalorraquídeo está comprendido entre 7,35 y 7,40. Aunque la sangre y otros fluidos disponen de un poder regulador importante que les permite tolerar la inyección de preparados con pH comprendidos entre 4 y 10, la administración de soluciones con valores de pH alejados del fisiológico puede producir dolor, así como inflamaciones y lesiones en tejidos y endotelios17.
2. Llenado
Se utilizan ampollas con una capacidad de 5 ml de vidrio transparente, cerradas, con banda autorrompible y estériles.
El llenado se realiza en un equipo BOSCH ARF 5020 (Figura 3). Se lleva a cabo sin introducir gas inerte (nitrógeno) en el interior de las ampollas, disponiendo así una atmósfera oxidativa y por ello favoreciendo los fenómenos oxidativos y la inestabilidad del fármaco, buscando el «peor caso».

Figura 3 Llenado de ampollas de ribavirina en la llenadora BOSCH ARF 5020 en el módulo de estériles del CEMILFARDEF.
Envasado de la Formulación 1: 211 ampollas con 5 ml/ampolla, de las cuales 105 se prepararon para pasar a la etapa de esterilización y 106 se prepararon para etiquetar.
Envasado de la Formulación 2: 233 ampollas con 5 ml/ampolla, de las cuales 100 se destinaron para pasar a la etapa de esterilización y 133 se prepararon para etiquetar.
3. Esterilización
Se realiza un ciclo de esterilización de 121 ºC - 20 minutos en un autoclave Celester Sterimega SM750, a un total 205 ampollas (105 de la F1 y 100 de la F2).
La esterilización terminal de un producto en su envase definitivo es prioritaria, salvo que se produzca una degradación significativa del producto, a causa de este proceso, que obligaría a trabajar en un proceso por vía aséptica17,18.
4. Etiquetado
Se etiquetan manualmente todas las ampollas, identificando fórmula, volumen, con/sin esterilización y fecha (Figura 4).

5. Método analítico
Se establecen los siguientes parámetros y ensayos para evaluar la calidad y estabilidad de la solución de Ribavirina al 10%: pH, riqueza mediante técnica Cromatografía de alta eficacia (HPLC) y ausencia de alteraciones físicas (turbidez, cambio de color, aparición de precipitado):
Valoración por HPLC (Agilent Technologies. Mod: 1260 Infinity): Se sigue el método indicado en monografía de la Farmacopea Europea16. Especificación: 90-110 mg/ml o 90-110%.
Medición pH de cada una de las muestras (pHmetro Mettler Toledo SEVEN COMPACT S220). Especificación: 4,5-6,0.
6. Estudio de estabilidad
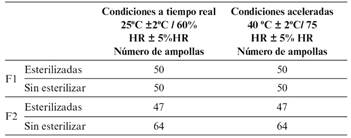
Las ampollas de cada una de las formulaciones fabricadas se separan en dos grupos (Tabla 5) para almacenarlas en condiciones a tiempo real (a largo plazo): 25ºC ± 2ºC/60% HR ± 5%HR; y en condiciones aceleradas (a corto plazo): 40ºC ± 2ºC/75 HR ± 5% HR; según ICH Q1A(R2)19, condiciones ambientales para zona climática I (templada) y II (subtropical), durante un tiempo de 6 meses para condiciones aceleradas y 12 meses para condiciones a tiempo real, y con estos últimos resultados se realiza una estimación estadística de un período de validez.
RESULTADOS
Todos los ensayos se realizaron en el departamento de Control de Calidad del CEMILFARDEF a los tiempos establecidos según ICH19:
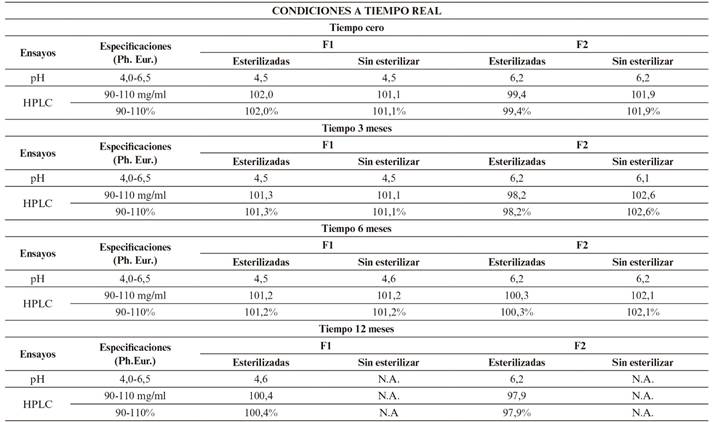
Resultados en condiciones a tiempo real (a largo plazo) según Tabla 6.
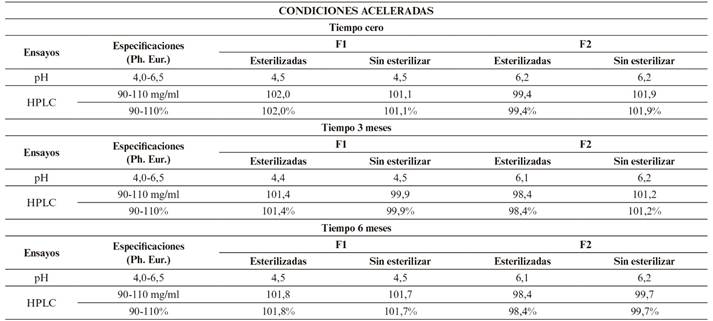
Resultados en condiciones aceleradas (a corto plazo) según Tabla 7.
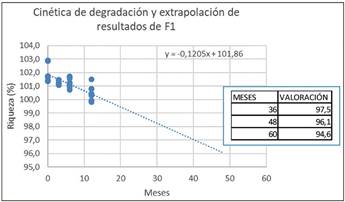
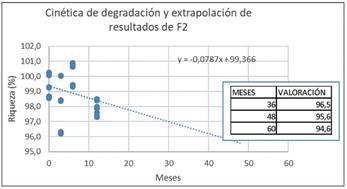
Extrapolación de los datos obtenidos a tiempo real en un estudio a 12 meses para la estimación de un posible periodo de validez (Figura 9 y Figura 10) en las fórmulas F1 y F2 esterilizadas.
DISCUSIÓN
Durante el estudio, tanto en condiciones a tiempo real como aceleradas, no se observan diferencias significativas entre los resultados obtenidos de pH o riqueza en cada una de las formulaciones (F1 y F2), tras haber sufrido un ciclo de esterilización, con respecto a las no esterilizadas. Por este motivo, después de trascurridos los 6 primeros meses, y comprobar que los parámetros de las muestras esterilizadas y no esterilizadas no presentan variaciones, se decide no continuar con los estudios de estabilidad de las muestras sin esterilizar y hacerlo únicamente con muestras esterilizadas, según se refleja tanto en la Tabla 6 como en la Figura 5 y 6.
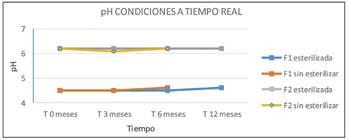

Los resultados obtenidos (Tabla 6 y 7) cumplen especificaciones de Farmacopea Europea para ambas formulaciones (F1 y F2), demostrando una estabilidad química tras su conservación en condiciones de estudio a largo plazo durante 12 meses y acelerados durante 6 meses. Por ello, tanto la formulación F1 como la F2 resultan adecuadas para su fabricación en un proceso industrial, pues su estabilidad química con respecto a su riqueza (Figura 6 y 8) y pH (Figura 5 y 7), mantienen valores incluidos en los límites de referencia establecidos para estas especificaciones.

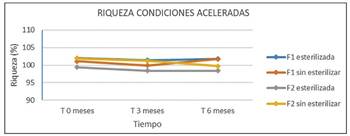
La estabilidad reflejada en condiciones aceleradas demuestra la robustez de la formulación y la ausencia de requerimientos especiales para su almacenamiento y transporte.
Ambas formulaciones no experimentan alteraciones en el valor de pH inicial durante el periodo de estudio, pero la Formulación 2 presenta un pH más próximo al sanguíneo/fisiológico, lo que evitará posibles dolores o lesiones en la zona de administración (Figuras 5 y 7), a pesar de que el proceso de fabricación de la Formulación 1 sea más simple al disponer una composición más sencilla.
Los resultados obtenidos en la evaluación de muestras a 12 meses, permiten establecer una estimación del contenido en sustancia activa. Como se observa en las Figuras 9 y 10, ninguna de las formulaciones supera el límite inferior establecido en la monografía de la ribavirina (Ph. Eur) del 90%, extrapolando a un período de 60 meses, en ambos casos se consigue un valor aproximado al 94,6% en condiciones de 25ºC ± 2ºC/60% HR ± 5%HR. Las rectas de regresión permiten predecir la estabilidad de la formulación durante un período de 60 meses.
CONCLUSIÓN
El desarrollo de la ribavirina 100 mg/ml (10% w/v) inyectable satisface una necesidad terapéutica relacionada con el tratamiento de primera elección en enfermedades víricas altamente contagiosas que carecen de tratamientos eficaces en Sanidad Militar.
Los ensayos efectuados confirman que el diseño de la solución es robusto, permanece estable en condiciones aceleradas y a tiempo real, y la solución tamponada presenta un pH compatible con sangre y tejidos.
Se propone completar los estudios de estabilidad (on going) según ICH Q1A(R2) durante 60 meses, para establecer un período de validez del medicamento y considerar este antiviral de amplio espectro en futuras revisiones del Petitorio de Farmacia Militar20 como medicamento de interés ante emergencias sanitarias provocadas por la dispersión de agentes víricos altamente infecciosos.
