










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkSESIÓN 2
8. Desarrollo y aplicación de secretoma con efecto osteogénico procedente de células madre mesenquimales para el tratamiento de la osteoporosis
González González A1, García Sánchez D1, Dotta M1, Reyes R2, Rodríguez Rey JC1, Pérez Campo FM1
1 Departamento de Bioquímica y Biología Molecular. Facultad de Medicina. Universidad de Cantabria-IDIVAL. Santander; 2 Departamento de Bioquímica, Microbiología, Biología Celular y Genética. Instituto de Tecnología Biomédica (ITB). Universidad de La Laguna
Introducción: Múltiples ensayos en modelos animales demuestran que la degeneración ósea ocurrida durante la osteoporosis puede potencialmente tratarse usando células madre mesenquimales (CMMs). Sin embargo, estas terapias no están exentas de problemas, lo que ha promovido la búsqueda de alternativas. Numerosas evidencias indican que la efectividad de las terapias con CMMs se debe principalmente a su actividad paracrina. Nuestro grupo ha demostrado que el silenciamiento transitorio en CMMs de un inhibidor de las rutas BMP (Smurf1) aumenta de forma significativa su capacidad osteogénica en modelos animales y en células de pacientes osteoporóticos. Nuestra hipótesis actual es que el silenciamiento de Smurf1 puede, además, incrementar el potencial osteogénico del secretoma producido por estas células, lo que permitiría usarlo a modo de fármaco, evitando las posibles complicaciones derivadas del trasplante de CMMs.
Material y método: Para producir el secretoma, se usó la línea de CMMs humanas ASC52telo. El silenciamiento de Smurf1, se realizó utilizando un GapmeR específico permitiendo posteriormente el crecimiento de las células durante 48 horas antes del aislamiento del secretoma (SECSmurf1). La capacidad osteogénica inducida por SECSmurf1 se ensayó in vitro en CMMs murinas y humanas, e in vivo, en un modelo ectópico murino. La valoración in vitro se llevó a cabo midiendo expresión de marcadores osteogénicos, actividad de la fosfatasa alcalina y grado de mineralización. Los ensayos in vivo se evaluaron mediante técnicas histológicas habituales.
Resultados: Los datos obtenidos en células primarias murinas muestran que el tratamiento con SECSmurf1 incrementa significativamente la capacidad osteogénica in vitro. Asimismo, los resultados in vivo, muestran un aumento en la presencia de núcleos de osificación, expresión de fosfatasa alcalina y osteocalcina en bioimplantes portadores de CMMs que han sido pre-tratadas con SECSmurf1. Estos resultados se mantienen al analizar el efecto de dicho secretoma sobre las CMMs aisladas de individuos osteoporóticos. Además, en estas células, la promoción de la capacidad osteogénica por el SECSmurf1 es equiparable a la obtenida en presencia de BMP2, el factor más usado actualmente en clínica para promover la regeneración ósea.
Conclusiones: En conjunto, este trabajo revela que el uso del secretoma SECSmurf1 puede ser una estrategia, libre de células, alternativa al uso directo de CMMs para el tratamiento de la osteoporosis.
9. Variantes genéticas como causa de hipofosfatemia de origen desconocido
Puente Ruiz N1, Vega Pajares AI1, García Unzueta MT1, Alio Lavin B1, Riancho Zarrabeitia L2, Mateos F2, Maiztegi A1, Valero Díaz de Lamadrid C1, Piedra León M1, Riancho JA1
1 Hospital Universitario Marqués de Valdecilla. Santander; 2 Hospital Sierrallana. Torrelavega
Introducción: En muchos pacientes con hipofosfatemia se detectan causas adquiridas, bien transitorias (como los trastornos agudos con redistribución al espacio intracelular -acidosis, síndromes linfoproliferativos) o bien persistentes, como la ingesta crónica de alcohol, el hiperparatiroidismo, los cuadros malabsortivos, o el uso de ciertos fármacos. Sin embargo, algunos pacientes presentan hipofosfatemia mantenida sin una causa secundaria evidente. Por eso, el objetivo de este estudio fue determinar el espectro etiológico de la hipofosfatemia persistente en pacientes adultos y su posible origen genético.
Material y métodos: Se exploraron las bases de datos del laboratorio del hospital para identificar los pacientes con hipofosfatemia. Se revisaron después sus historias en busca de causas adquiridas de hipofosfatemia. Los pacientes sin causa evidente fueron invitados a acudir a una consulta para un estudio clínico y bioquímico. En caso de no identificarse causa, se hizo un análisis molecular, mediante secuenciación (NGS) de las regiones exónicas e intrónicas flanqueantes de un panel de genes relacionados con raquitismo o hipofosfatemia (CLCN5, CYP27B1, DMP1, ENPP1, FAM20C, FGFR1, FGF23, GNAS, PHEX, SLC34A3 y VDR).
Resultados: Entre los 284 pacientes con hipofosfatemia persistente que pudieron ser estudiados, las causas secundarias más frecuentes fueron: hiperparatiroidismo primario (17%), alcohol (15%), hipovitaminosis D (15%), trasplante (10%), tratamiento antirretroviral (9%), cirugía bariátirca (8%), tratamiento con hierro intravenoso (5%) y trastornos de la conducta alimentaria (2%).
En 39 pacientes (14%), en los que no había una causa adquirida, incluyendo 2 casos diagnosticados de raquitismo hipofosfatémico ligado al cromosoma X, se realizó el estudio genético. En 12 de estos (31%) se encontró una variante potencialmente patogénica en alguno de los genes estudiados (tabla).
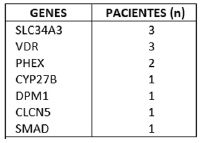
Conclusiones: Se deben incluir las causas genéticas dentro del diagnóstico diferencial de las hipofosfatemias persistentes de origen desconocido, que pueden explicar casi un tercio de los casos de los pacientes adultos.
10. Influencia del confinamiento por COVID-19 sobre la incidencia y mortalidad de las fracturas mayores osteoporóticas
Surís X1, Vela E2, Clèries M2, Llargués E3, Camins J3, Larrosa M1
1 Plan director de enfermedades reumáticas y del aparato locomotor. CatSalut. Hospital General de Granollers; 2 Unitat de Infirmació i Coneixement. CatSalut; 3 Hospital General de Granollers
Introducción: El propósito del estudio fue analizar el impacto del confinamiento por COVID-19 sobre la incidencia y mortalidad de las fracturas mayores osteoporóticas (FMO) durante 2020.
Material y métodos: Se obtuvieron diagnósticos de fracturas incidentes en ≥ 50 años entre 2018 y 2020 a través del Registro de Morbilidad y Utilización de Recursos Sanitarios de Cataluña. Se analizaron los diagnósticos de hospitalización, atención urgente y atención primaria. Se estimaron las tasas para FMO por tramos de edad, sexo y comorbilidades. Las tasas diarias de fractura y la mortalidad en 2020 y en los dos años anteriores se compararon en cuatro períodos de tiempo: 01/01 al 14/03 (preconfinamiento), 15/03 al 28/04 (confinamiento estricto), 29/04 al 28/06 (desconfinamiento) y de 29/06 en adelante (posconfinamiento).
Resultados: De enero del 2018 a noviembre del 2020 se registraron 88.722 FMO: 57.722 en mujeres (edad media 79 años) y 18.509 en hombres (edad media 76 años). La tasa de incidencia anual fue de 10,4 para el total de fracturas. Las tasas de fractura disminuyeron durante el confinamiento estricto [RR 0,57 (0,52-0,63) en hombres y 0,56 (0,53-0,60) en mujeres, p>0,001] y permanecieron más bajas durante el resto del año (figura). La disminución fue mayor en los grupos de edad más jóvenes y sanos, en las fracturas vertebrales, pélvicas y de brazo. La fractura de cadera disminuyó de manera significativa, pero con menor intensidad [RR 0,87 (0,8-0,94), p<0,001]. La mortalidad posfractura aumentó a lo largo de 2020 hasta 2,5 veces durante el confinamiento. El exceso de mortalidad estuvo relacionado con COVID-19.
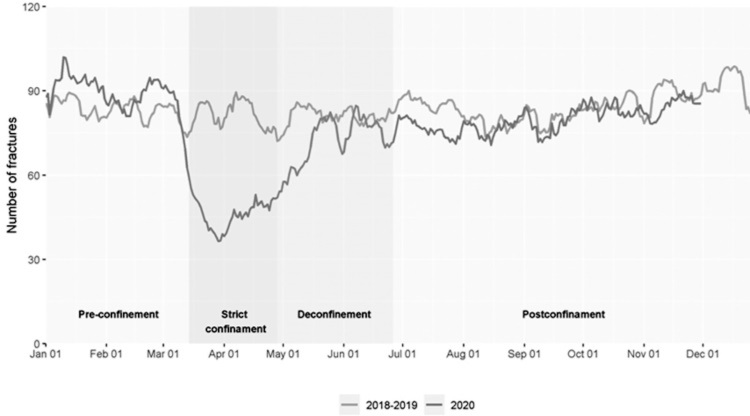
Conclusiones: El confinamiento por COVID-19 se asoció con una reducción de la incidencia de FMO, especialmente en personas más jóvenes y en fracturas distintas de la cadera. La mortalidad posfractura fue mayor que en años anteriores debido a la alta mortalidad por COVID19 en los ancianos.
11. Caracterización funcional y estructural de nuevas variantes del gen ALPL asociadas a hipofosfatasia
García Fontana B1, Jiménez Ortas A2, Martínez Heredia L3, Andújar Vera F4, Sanabria de la Torre R3, González Salvatierra S3, Andreo López MC5, Cabrera Gómez N3, Muñoz Torres M6, García Fontana C6
1 Unidad de Endocrinología y Nutrición. Hospital Universitario Clínico San Cecilio de Granada. Instituto de Investigación Biosanitaria de Granada (Ibs.GRANADA). CIBERFES. Instituto de Salud Carlos III; 2 Dpto. Bioquímica y Biología Molecular II, Universidad de Granada; 3 Dpto. de Medicina. Universidad de Granada. ibs.GRANADA; 4 ibs.GRANADA. Departamento de Informática e Inteligencia Artificial. Universidad de Granada. Instituto Andaluz de Investigación en Ciencia de Datos e Inteligencia Computacional (Instituto DaSCI); 5 Unidad de Endocrinología y Nutrición. Hospital Universitario Clínico San Cecilio. Granada; 6 Unidad de Endocrinología y Nutrición. Hospital Universitario Clínico San Cecilio de Granada. Ibs.GRANADA. CIBERFES. Instituto de Salud Carlos III; Dpto. de Medicina. Universidad de Granada
Introducción: La hipofosfatasia (HPP) es una enfermedad genéticararacaracterizada por una disminución de la actividad enzimática de la fosfatasa alcalina no específica de tejido (TNSALP)a causa de mutaciones en el gen ALPL que codifica esta proteína.Aunquelas principales manifestaciones clínicas de HPP ocurrena nivel óseo, existe una gran heterogeneidad clínica que frecuentemente dificulta el diagnóstico debido al solapamiento de su sintomatología con la de otras patologías más prevalentes
Objetivos: Caracterización funcional y estructural de dos nuevas variantes en el gen ALPL no descritas previamente (p.Leu6Ser y p.Thr167del) y estudio de su relación con las manifestaciones clínicas para tratar de establecer una relación geno-fenotípica en cada una de las variantes.
Métodos: Se transfectaron células HEK293T con el vector pcDNA 3.1 vacío y conteniendo la secuencia codificante de la proteína wild type (WT) y de cada una de las mutaciones del gen ALPL objeto de estudio.La eficiencia de la transfección se comprobó mediante qPCR. Se determinó la actividad enzimática de las proteínas codificadas por las distintas variantes mediante Alkaline Phosphatase Assay Kit (Abnova) y se realizó un modelado tridimensional de las mismas con AlphaFold para analizar el efecto de las mutaciones a nivel estructural.
Resultados: Ambas variantes mostraron una actividad enzimática significativamente disminuida respecto a la proteína WT (p<0,001), presentando la variantep.Thr167del los valores más disminuidos. Los ensayos de modelaje 3D mostraron una significativa reducción del tamaño del sitio activo de la proteína en el caso de la variante pThr167del mientras que la variante p.Leu6Ser no mostró alteraciones significativas a nivel estructural. En cuanto a sintomatología, el paciente portador de la variante p.Leu6Ser presentó sólo molestas musculares mientras que el paciente portador de la variante p.Thr167del mostró notables problemas digestivos y psoriasis.
Conclusiones: Nuestros resultados sugieren que la variante p.Leu6Ser se asocia a un fenotipo leve de HPP, mientras que la variante p.Thr167del se asocia a un fenotipo moderado de HPP.
12. Análisis de los mediadores del metabolismo óseo en pacientes con artritis reumatoide de inicio en el anciano, y su relación con la actividad de la enfermedad y la masa ósea
Brandy García AM1, Martínez Morillo M2, Cora R3, Prior Español A2, Mateo L2 , Guma M4, Gifre L2
1 Hospital Universitario Cabueñes/Hospital Universitario Germans Trias i Pujol. Badalona; 2 Hospital Universitario Germans Trias i Pujol. Badalona; 3 Department of Medicine, School of Medicine, University of California, San Diego (USA); 4 Department of Medicine, School of Medicine, University of California, San Diego (USA)/Departmento de Medicina, Universidad Autónoma de Barcelona
Introducción: Los pacientes con artritis reumatoide de inicio en el anciano (EORA) presentan síntomas más agudos, mayor elevación de reactantes de fase aguda (RFA) y reciben mayores dosis de corticoides (GC) que los pacientes más jóvenes. El objetivo de este estudio es analizar los mediadores del remodelado óseo (Dkk-1, SOST, OPG y RANKL) en estos pacientes y su relación con la actividad de la enfermedad y la masa ósea.
Material y métodos: Estudio observacional prospectivo en el que se incluyeron pacientes con EORA sin osteoporosis conocida. Se recogieron datos clínicos, analíticos y los mediadores del remodelado óseo al inicio (sin tratamiento) y a los 1, 3 y 12 meses. La DMO y sCTX se evaluaron basal y al año. Los resultados se compararon con un grupo control.
Resultados: Se incluyeron 42 pacientes con EORA (18M:24H) con media de edad 74±7 años. Los pacientes con EORA presentaban mayores valores de OPG y SOST que el grupo control, que se mantuvieron elevados durante el seguimiento; mientras que Dkk-1 estaba disminuido (figura). sCTX estaba aumentado en el momento basal, normalizándose al año. Se observó una correlación positiva entre SOST y los RFA en el momento basal; sin embargo, no se observaron otras correlaciones entre mediadores óseos, parámetros de actividad de la enfermedad o la dosis de GC. Densitométricamente, el 28,6% de los EORA tenía una osteoporosis basal; y tanto OPG como sCTX se correlacionaron negativamente con la DMO. Destacar que sCTX mostró una buena capacidad discriminativa para detectar la presencia de osteoporosis. A los 12 meses, presentaron una discreta pérdida de masa ósea femoral.
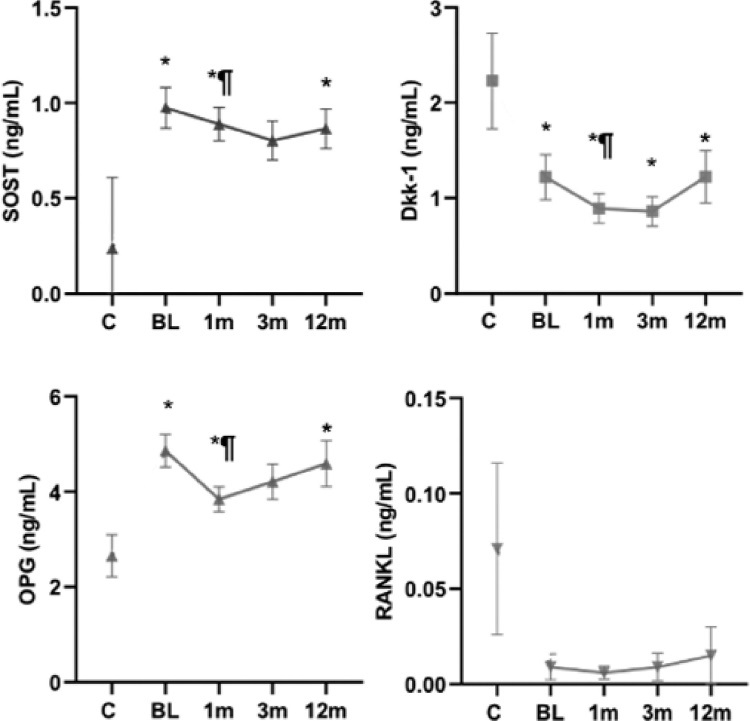
Evolución de mediadores óseos en pacientes con EORA y grupo de control. * p<0.05 comparado con controles. ¶ p<0.05 comparado con EORA basal tras corrección de Bonferroni.
Conclusiones: Los mediadores óseos en los pacientes con EORA se relacionan con los parámetrosde inflamación, especialmente SOST, y con la enfermedad por sí misma. sCTX parece ser un marcador útil para discriminar la presencia de osteoporosis en EORA.
13. Experiencia colaborativa en una Unidad Multidisciplinar de Displasias Esqueléticas
Tornero C1, Heath KH2, García Carazo S1, Monjo Henry I3, Bernad Pineda M3, Fernández E3, Balsa A3, Aguado P1
1 Servicio de Reumatología. Hospital Universitario La Paz y Unidad Multidisciplinar de Displasias Esqueléticas (UMDE), Hospital Universitario La Paz. Madrid; 2 Instituto de Genética Médica y Molecular (IN-GEMM). Hospital Universitario La Paz y CIBERER, ISCIII. Unidad Multidisciplinar de Displasias Esqueléticas (UMDE). Hospital Universitario La Paz. Madrid; 3 Servicio de Reumatología. Hospital Universitario La Paz. Madrid
Introducción: La creación de unidades multidisciplinares de displasias esqueléticas, generalmente constituidas por pediatras y especialistas en genética, ha contribuido a un mejor abordaje de las enfermedades minoritarias en edad infantil. Sin embargo, la integración de especialistas en adultos es menos frecuente, y la transición a edad adulta una necesidad a menudo no cubierta. En nuestro hospital terciario, la experiencia colaborativa de la Unidad Metabólica Ósea (UMO) de Reumatología en la Unidad Multidisciplinar de displasias esqueléticas (UMDE) del centro se inició en 2019 tras establecerse en nuestro servicio una línea de investigación en el campo de la hipofosfatasia (HPP) del adulto que fomentó la derivación de pacientes con otros tipos de displasia. El objetivo de este trabajo consiste en analizar los diagnósticos de los adultos con displasias esqueléticas atendidos en nuestra UMO en el seno de esta experiencia colaborativa.
Material y métodos: Análisis trasversal descriptivo de diagnósticos y estudios genéticos de adultos que de manera consecutiva fueron derivados para evaluación de displasia ósea en la UMO de Reumatología durante el período 01/2019-05/2022. La procedencia de los pacientes incluyó los servicios de Genética Clínica, Endocrinología Infantil, otros centros hospitalarios de la Comunidad de Madrid y pacientes derivados a consultas de Reumatología desde el área de salud de nuestra influencia.
Resultados: Se estudiaron 65 pacientes con una edad media de 51,6±16 (69% mujeres). Cuarenta y dos pacientes presentaron mutaciones en el gen ALPL (40 con manifestaciones compatibles con hipofosfatasia y 2 portadores); 9 pacientes presentaron osteogénesis imperfecta; 3 con osteoporosis y múltiples fracturas en la postmenopausia reciente, mostraron variantes patogénicas en el gen WNT1; 2 pacientes, raquitismo hipofosfatémico ligado al X (variante en gen PHEX) y las siguientes displasias estuvieron presentes cada una en un paciente: osteopetrosis (variante en gen CLCN7), sinostosis múltiple tipo I, (variante en gen NOG), síndrome de Lenz, síndrome de Rubinstein Taybi, síndrome de VAC-TERL, síndrome de Stickler tipo 2, síndrome de Turner, displasia acromícrica (variante en gen FNB1) y osteopoiquilia.
Conclusión: La participación del reumatólogo y otros especialistas del metabolismo óseo en adultos resulta fundamental para una adecuada atención de pacientes con enfermedades minoritarias óseas , tanto para su diagnóstico precoz como para el seguimiento en la transición desde la infancia. La integración de nuestra UMO en la UMDE de nuestro hospital ha dado lugar a una actitud derivativa proactiva de casos de pacientes en edad adulta.

