










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.106 no.5 Madrid may. 2014
CARTAS AL EDITOR
Aceruloplasminemia: una entidad a considerar en pacientes con anemia ferropénica
Aceruloplasminemia: An entity to consider in patients with anemia
Palabras clave: Aceruloplasminemia. Ceruloplasmina. Anemia. Ferritina. Hiperferritinemia. Sobrecarga férrica. Hemocromatosis. Quelantes del hierro. Deferoxamina.
Key words: Aceruloplasminemia. Ceruloplasmin. Anemia. Ferritin. Hyperferritinemia. Iron overload. Hemochromatosis. Iron chelator. Desferrioxamine.
Sr. Editor:
La aceruloplasminemia es una entidad genética excepcional en nuestro medio pero que comparte características clínicas y analíticas con enfermedades propias de la gastroenterología como la hemocromatosis, la enfermedad de Wilson o la enfermedad de Menkes. Además, el primer signo de la enfermedad es la anemia, frecuente motivo de consulta en nuestra especialidad.
Caso clínico
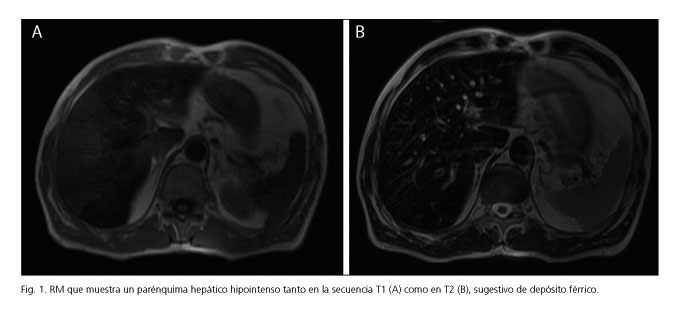
Presentamos un caso de un varón de 47 años remitido por dolor abdominal y anemia (Hb 11,4 mg/dl y hierro sérico 15 µg/dl) asociada a hiperferritinemia (707 ng/ml). La gastroscopia, colonoscopia, ecografía y tránsito gastrointestinal descartaron afectación inflamatoria. A pesar de presentar un índice de saturación de la transferrina bajo (3,7 %) se realizó estudio genético de hemocromatosis que fue negativo. Dada la normalidad de las pruebas se solicitó la ceruloplasmina y estudio del metabolismo del cobre, que confirmaron el diagnóstico de aceruloplasminemia: ceruloplasmina inferior a 7 mg/dl (rango normal, 22-61 mg/dl), cobre sérico 13 µg/dl (rango normal, 70-150 µg/dl) y cobre en orina de 24 horas normal. No se realizó test genético por no encontrarse disponible en nuestro centro. La resonancia magnética (RM) abdominal identificó baja intensidad de señal en T1 y T2 compatible con depósito hepático de hierro (Fig. 1) sin observar alteraciones en la RM cerebral ni en el estudio oftalmológico.
Discusión
La ceruloplasmina, enzima con actividad ferroxidasa, permite la incorporación del hierro a la transferrina y su movilización desde los tejidos (1-3). La mutación del gen CP conduce a una acumulación de hierro en el hígado, páncreas y el sistema nervioso central (4). La acumulación en el hígado no conduce al desarrollo de daño hepático (5). Existen unas 40 mutaciones descritas, pero su determinación está limitada debido al gran tamaño de la región codificada (6,7). Es conveniente ofrecer consejo genético a los afectados y familiares (5).
Clínicamente cursa con degeneración retiniana, diabetes mellitus y alteraciones neurológicas pero frecuentemente la anemia antecede a estas manifestaciones (5,8). Se presenta en la cuarta o quinta década de la vida y la muerte suele producirse alrededor de los 65 años (1,5,8).
Analíticamente se caracteriza por ausencia total o casi total de ceruloplasmina, niveles de cobre y hierro sérico bajos con ferritina elevada y actividad ferroxidasa indetectable (1,9).
Es importante realizar el diagnóstico diferencial con otras enfermedades que cursan con hiperferritinemia, como el síndrome metabólico, condiciones inflamatorias, el alcoholismo o la hemocromatosis y debe sospecharse ante la presencia de anemia una vez descartadas las causas más frecuentes (5). Los niveles séricos de ceruloplasmina también se encuentran disminuidos en los trastornos del metabolismo del cobre, por lo que debe diferenciarse de la enfermedad de Wilson, en la que existe incapacidad para transferir el cobre al precursor de la ceruloplasmina y de la enfermedad de Menkes, en la que la absorción intestinal de cobre está reducida y por tanto la síntesis de ceruloplasmina (5).
El tratamiento con quelantes del hierro está indicado en pacientes sintomáticos con cifras de hemoglobina superiores a 9 g/dl, aunque su eficacia para disminuir el depósito de hierro a nivel cerebral es escasa. La administración reiterada de plasma fresco congelado con ceruloplasmina podría mejorar los síntomas neurológicos (5). Las flebotomías están limitadas por la anemia asociada a esta entidad y pueden empeorar el cuadro clínico (9).
En resumen, se trata de una entidad a tener en cuenta ante la presencia de hiperferritinemia o anemia ya que su diagnóstico tiene importantes implicaciones pronósticas.
María Lozano Varela, Elisa Carrera Alonso y Gema Plaza Palacios
Servicio de Aparato Digestivo. Hospital Universitario de Guadalajara. Guadalajara
Bibliografía
1. Pérez-Aguilar F. Ceruloplasmina y metabolismo del hierro: sus implicaciones en la hemocromatosis, la enfermedad de Wilson y la aceruloplasminemia. Rev Clin Esp 2002;202:649-51. [ Links ]
2. Kono S, Miyajima H. Molecular and pathological basis of aceruloplasminemia. Biol Res 2006;39:15-23. [ Links ]
3. Harris ZL, Takahashi Y, Miyajima H, Serizawa M, MacGillivray RT, Gitlin JD. Aceruloplasminemia: Molecular characterization of this disorder of iron metabolism. Proc Natl Acad Sci USA 1995;92:2539-43. [ Links ]
4. Finkenstedt A, Wolf E, Höfner E, Gasser BI, Bösch S, Bakry R, et al. Hepatic but not brain iron is rapidly chelated by deferasirox in aceruloplasminemia due to a novel gene mutation. J Hepatol 2010;53:1101-7. [ Links ]
5. Miyajima H. Aceruloplasminemia. GeneReviews. Available at: http//www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=acp (15.05.2008). [ Links ]
6. Fasano A, Colosimo C, Miyajima H, Tonali PA, Re TJ, Bentivoglio AR. Aceruloplasminemia: A novel mutation in a family with marked phenotypic variability. Mov Disord 2008;23:751-5. [ Links ]
7. Kono S, Suzuki H, Oda T, Shirakawa K, Takahashi Y, Kitagawa M, et al. Cys-881 is essential for the trafficking and secretion of truncated mutant ceruloplasmin in aceruloplasminemia. J Hepatol 2007;47:844-50. [ Links ]
8. McNeill A, Pandolfo M, Kuhn J, Shang H, Miyajima H. The neurological presentation of ceruloplasmin gene mutations. Eur Neurol 2008;60:200-5. [ Links ]
9. Bethlehem C, Van Harten B, Hoogendoorn M. Central nervous system involvement in a rare genetic iron overload disorder. Neth J Med 2010;68:316-8. [ Links ]

