










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkAnales de Medicina Interna
versão impressa ISSN 0212-7199
An. Med. Interna (Madrid) vol.20 no.12 Dez. 2003
| Estudio de calidad del diagnóstico y seguimiento de los trastornos esqueléticos asociados a la enfermedad de Gaucher tipo 1 en España J. I. PÉREZ CALVO, P. GIRALDO CASTELLANO1, D. RUBIO-FÉLIX1, M. GIRALT REICHS1, M. POCOVÍ MIERAS2 Servicio de Medicina Interna. Hospital Clínico Universitario Lozano Blesa. 1Servicio de Hematología. Hospital Universitario Miguel Servet. 2Departamento de Bioquímica, Biología Molecular y Celular. Facultad de Ciencias. Universidad de Zaragoza. Zaragoza
|
| QUALITY STUDY OF DIAGNOSTIC AND FOLLOW-UP PROCEDURES OF BONE INVOLVEMENT IN TYPE 1 GAUCHER'S DISEASE IN SPAIN
| ||
| RESUMEN Introducción: La afectación ósea de la enfermedad de Gaucher (EG) tipo 1 es invalidante y silente, para su diagnóstico debe realizarse resonancia magnética (RM) sistemáticamente. El objetivo del estudio fue saber si el tipo de hospital implica diferencias en el uso de medios diagnósticos en la EG. Material y métodos: Se analizan los recursos diagnósticos disponibles y su empleo en la afectación ósea según el tipo de hospital. Resultados: Treinta y un pacientes de hospitales de referencia y 16 de comarcales. El 70% de los casos presentaban afectación ósea (formas graves el 60%). La RM se empleó en el diagnóstico inicial (65%), y el seguimiento (93%) especialmente en hospitales de referencia. La determinación de quitotriosidasa se emplea el 60% de los casos en seguimiento. Conclusiones: La red de hospitales públicos españoles está suficientemente dotada para la atención de pacientes con EG en todos sus niveles asistenciales, aunque se infrautilizan los recursos disponibles. PALABRAS CLAVE: Enfermedad de Gaucher. Tesaurismosis. Enfermedad ósea. Procedimientos diagnósticos. Resonancia magnética. Quitotriosidasa. | ABSTRACT Background: bone involvement in type 1 Gaucher’s disease can be devastating and is oftenly silent. Bone MR is generally recomended. The aim of the study was to elucidate whether the size and location of hospitals in Spain implies any difference in management of GD patients. Material and methods: we surveyed the type of facilities in the hospital (namely MRI) as well as for the presence, type, severity and methodology of follow-up of bone involvement associated to GD, according to the category of hospital (local or reference). Results: 31 patients were followed in reference hospitals whereas 16 other were in local hospitals. 70% of cases had some type of bone involvement, 60% had severe bone disease. MRI was the first choice for diagnosis in 65% and for follow-up in 93% of cases. MRI is less indicated among patients from local hospitals. Chitotriosidase is measured in a high, but insufficient, proportion of the followed patients (60%). Conclusions: the Spanish hospital network, either reference or local hospitals, have an adequate infrastructure for the management of GD patients. However, main diagnostic resources are being currently underused. KEY WORDS: Gaucher disease. Tesaurismosis. Bone disease. Management. Magnetic resonance imaging. Chitotriosidase. |
Pérez Calvo JI, Giraldo Castellano P, Rubio-Félix D, Giralt Reichs M, Pocoví Mieras M. Estudio de calidad del diagnóstico y seguimiento de los trastornos esqueléticos asociados a la enfermedad de Gaucher tipo 1 en España. An Med Interna (Madrid) 2003; 20: 607-611.
Trabajo aceptado: 14 de mayo de 2003
Correspondencia: Juan I. Pérez Calvo. Servicio de Medicina Interna. Hospital Clínico Universitario Lozano Blesa. Avda S. Juan Bosco, 15. 50009 Zaragoza. e-mail: mibh-jperez@hcu-lblesa.es
INTRODUCCIÓN
La enfermedad de Gaucher (EG) es una enfermedad de etiología genética, con herencia autosómica recesiva, producida por el déficit enzimático de beta-glucosidasa ácida o cerebrosidasa, responsable de la hidrólisis intracelular de la glucosil-ceramida y otros glucoesfingolípidos afines. Desde el punto de vista genético el trastorno es secundario a mutaciones del gen que la codifica , localizado en el cromosoma 1 (1 q 21) (1,2).
La EG es la más frecuente de la enfermedades lisosomales, afecta sujetos de todas las razas y su frecuencia es de 1/40.000-60.000 en la población general (3). En España estimamos una frecuencia ligeramente inferior, de alrededor de 1/100.000 (4).
Al ser la ß-glucosidasa ácida una enzima de origen lisosomal macrofágica, la expresividad clínica es mayor en aquellos órganos ricos en macrófagos, como la médula ósea, el bazo, el hígado (5) y el pulmón (6). Desde el punto de vita clínico la EG se clasifica en tres tipos, según el SNC esté indemne (tipo 1), o no (tipos 2 y 3). La primera permite supervivencias prolongadas o normales, mientras que las últimas ocasionan la muerte en la infancia o juventud respectivamente. De estas manifestaciones, la afectación ósea, además de muy frecuente (7), es la más impredecible ya que puede presentarse de modo súbito y producir complicaciones invalidantes e irreversibles (8). Por ello se recomienda realizar ya en el momento del diagnóstico inicial y con periodicidad anual en el seguimiento, un profundo examen óseo, mediante resonancia magnética (RM) independientemente de la sintomatología (9).
En la actualidad es posible realizar en España, de manera uniforme y centralizada, con un protocolo único, el diagnóstico genético y el seguimiento bioquímico de los pacientes por cualquier facultativo que lo solicite, gracias al servicio que presta la Fundación Española para el Estudio y Terapéutica de la Enfermedad de Gaucher (FEETEG). Sin embargo, la afectación ósea, que escapa al examen clínico, debe investigarse de modo sistemático y específico mediante RM, lo que impide que se lleve a cabo centralizadamente y ha de realizarse en cada uno de los centros donde el paciente es diagnosticado inicialmente.
Un buen número de pacientes afectos de EG en España son seguidos en hospitales comarcales, lo que junto a la necesidad de exploraciones sofisticadas para la evaluación inicial y el seguimiento de la enfermedad nos llevó a preguntarnos si existirían diferencias en el empleo de métodos diagnósticos y, por ende, en el tratamiento entre los pacientes con EG atendidos en hospitales comarcales y aquellos procedentes de hospitales de referencia.
MATERIAL Y MÉTODOS
Hemos realizado una encuesta por correo postal entre los médicos de toda España, de hospitales comarcales y de referencia, responsables de pacientes con EG. En ella se requería información sobre la especialidad del médico, el tamaño, dotación en recursos del centro y la distancia al centro de referencia. Además se solicitaba información sobre si cada paciente padecía o no manifestaciones óseas y el tipo de las mismas, la técnica empleada en el diagnóstico inicial y en el seguimiento evolutivo. Finalmente se solicitó información sobre el tratamiento de sustitución enzimática (TSE), sus dosis y la respuesta al mismo (Tabla I).

Sobre la dotación de recursos diagnósticos en el hospital se solicitó información específica acerca de si contaban en cada centro con aparataje para ecografía, tomografía computarizada (TC), RM, densitometría ósea, laboratorio de medicina nuclear, radiología vascular y/o intervensionista, banco de sangre y laboratorio de inmunología.
En cuanto al tipo de trastornos óseos se requirió información concreta sobre la presencia o ausencia los siguientes –de gravedad creciente– expansión de la cavidad medular diafisaria de los huesos largos (deformidad en matraz de Erlenmeyer); osteopenia; osteolisis; osteonecrosis; osteoesclerosis; afectación articular y si era portador de prótesis articular.
Se solicitaba información sobre la metodología empleada en el diagnóstico inicial de la afectación ósea y el seguimiento de la EG globalmente y de la afectación ósea en particular, considerando las siguientes posibilidades: diagnóstico por la clínica, ultrasonografía y/o TC, determinación de quitotriosidasa plasmática (QT) y RM.
Finalmente, sobre el tratamiento se preguntaba por la causa que motivó la indicación, la dosis total mensual, la pauta de administración (quincenal o semanal) y la respuesta al mismo (aparición de nuevos episodios, estabilización –lesiones residuales sin aparición de otras nuevas– o regresión de lesiones anteriores).
RESULTADOS
Se enviaron un total de 68 encuestas de las que se contestaron 28 (41,1%), conteniendo información de 47 pacientes (22 varones y 25 mujeres) pertenecientes a 34 familias no relacionadas. De estos, 31 pacientes procedían de hospitales de referencia y 16 de comarcales.
Los pacientes de esta muestra son atendidos por especialistas en Medicina Interna (16 pacientes), Hematología (19 pacientes), Pediatría (10 pacientes) y Aparato Digestivo (1 paciente).
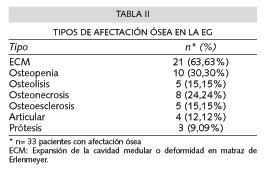
AFECTACIÓN ÓSEA EN LA EG
En la presente cohorte, 33 pacientes de los 47 registrados presentan afectación ósea en el contexto de su EG, lo que supone un 70,21% de los casos.
El tipo de afectación más frecuente es la deformidad en matraz de Erlenmeyer, la forma más leve y más común, generalmente combinada con otras. No obstante, la presencia de formas graves, como la osteonecrosis, osteoesclerosis o afectación articular con o sin implantación protésica afecta a un porcentaje nada despreciable alrededor del 60% de los casos (Tabla II).
EMPLEO DE RECURSOS CLÍNICOS Y COMPLEMENTARIOS EN LA EG
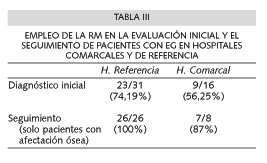
La dotación en tecnología diagnóstica difiere entre los hospitales de referencia y los comarcales en aspectos como la radiología vascular o intervensionista o la densitometría ósea con la que ningún hospital comarcal de este grupo cuenta. Sin embargo todos los hospitales encuestados, salvo un comarcal, cuentan con salas de RM.
La RM es frecuentemente empleada en la evaluación de la afectación ósea, tanto en el momento del diagnóstico inicial (65% de los pacientes), como en el seguimiento de los pacientes, especialmente los que están en tratamiento substitutivo (93% de los casos). Sin embargo su empleo difiere en los hospitales de referencia y en los comarcales especialmente en cuanto a su uso en la evaluación inicial de los pacientes (Tabla III).
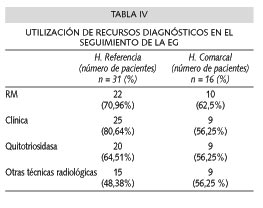
En el control evolutivo de las manifestaciones generales, distintas de las óseas, de los pacientes con EG se emplean recursos de tipo clínico, bioquímico y radiológico (Tabla IV).
TRATAMIENTO DE LA EG
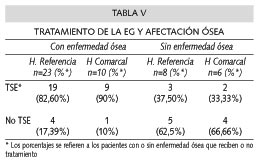
El 70% del total de los pacientes de esta serie y alrededor del 85% de los que padecen afectación ósea se encuentran en tratamiento de sustitución enzimática (TSE) (Tabla V). Estos datos son superponibles al conjunto de los pacientes españoles.
Las indicaciones que motivaron más frecuentemente el inicio de la terapia fueron (únicas o asociadas) las visceromegalias, las citopenias sanguíneas, y las alteraciones óseas por este orden.
Todos los pacientes se tratan con esquemas de dosis intermedias (30 U/k) o altas (60 U/k) quincenales, menos un paciente que recibe 2,3 U/k 3 veces por semana.
En 10 pacientes las lesiones óseas se estabilizaron tras el inicio de la TSE, en 6 se obtuvo una regresión de las mismas y en 2 se detectó progresión a pesar del tratamiento. Estos dos últimos recibían dosis quincenales, que se aumentaron.
DISCUSIÓN
En el informe del International Collaborative Group on Gaucher Disease (ICGGD) de 1994 centrado en los aspectos epidemiológicos de la afectación ósea de la EG se ponían de manifiesto aspectos muy relevantes de esta. En primer lugar su alta frecuencia, ya que un 88% de los pacientes la presentaban. Además, el porcentaje de pacientes con afectación esquelética que no presentaban signos de afectación hematológica, o estos eran mínimos, alcanzaba el 20% y de ellos, más de las tres cuartas partes sufrían formas de enfermedad esquelética consideradas graves. A pesar de estos datos, la afectación esquelética acostumbra a ser paucisintomática en el doble sentido del término. Por una parte se expresa por pocos síntomas o signos, generamente dolor y, ocasionalmente fiebre, y además son pocos los pacientes sintomáticos.
Por lo tanto la afectación ósea en la EG es frecuente, frecuentemente grave, oligosintomática y además impredecible. Por estas razones en los últimos años ha despertado un creciente interés su diagnóstico, estadíaje y tratamiento. Tanto es así que, en el momento presente se considera la exploración mediante RM del esqueleto como un examen básico, obligado, en todo paciente con EG (10). Además, el hallazgo de una afectación ósea extensa, aun siendo asintomática, se considera criterio suficiente para iniciar la TES (11).
En esta serie, en concordancia con los datos referidos la afectación ósea es mayoritaria, entre los pacientes de EG, puesto que esta presente en el 70% de los casos. La deformidad en matraz de Erlenmeyer es el hallazgo más común, pero es muy llamativo el porcentaje de pacientes con formas óseas graves, es decir, con necrosis diafisaria o de superficies articulares, que es superior al 60%. Esto quiere decir que además de frecuente la EG a nivel óseo es grave y puede adquirir un carácter muy invalidante. De acuerdo con lo anterior no es sorprendente que la mayoría de los pacientes con afectación ósea estén recibiendo TES y, al contrario, la mayoría de enfermos sin trastornos esqueléticos no son subsidiarios de la misma (Tabla V).
Afortunadamente, y en contra de nuestra hipótesis de trabajo, que preveía una mayor precariedad entre los pacientes tratados en hospitales pequeños, por una hipotética inferioridad en cuanto a su dotación tecnológica, esta es suficiente con independencia del medio hospitalario. Sin embargo, si existen algunas diferencias en cuanto al empleo de la RM. En los pacientes procedentes de hospitales comarcales el diagnóstico inicial de la enfermedad ósea, mediante RM solo alcanza a un 56%, frente al 74% de los hospitales de referencia. Esta situación cambia en el periodo de seguimiento en el que la RM se emplea de modo prácticamente sistemático en ambos. Esta paradoja, es decir la no utilización de una técnica tan informativa aun disponiendo de ella, es más aparente que real. Hasta la disponibilidad de un tratamiento eficaz para la EG en España en 1993 (12) había un buen número de pacientes diagnosticados, tanto en hospitales de referencia, como comarcales. En una buena parte de ellos se inicio el tratamiento ya en aquella época, en la que probablemente no existía la misma disponibilidad de infraestructuras en los hospitales comarcales con respecto a los de referencia, y la importancia y características de los trastornos óseos todavía no eran bien conocidos. Todos esos pacientes iniciaron el tratamiento sin tener una evaluación basal de su enfermedad ósea mediante RM, la cual ha ido realizandose a lo largo del seguimiento.
Nuestros resultados ponen de manifiesto la existencia en el momento actual de una red sanitaria pública con una cartera de servicios excelente y sobradamente dotada para llevar a cabo el control y tratamiento de los pacientes con EG en condiciones óptimas y similares en cualquier centro.
Caben, sin embargo, comentarios de matiz a esta afirmación. A pesar de su disponibilidad, prácticamente universal en esta muestra, la RM es una técnica infrautilizada, al menos en comparación a una situación ideal. Se usa en la evaluación inicial de la afectación ósea entre el 56 y el 74%, lo que puede conducir a subestimar la enfermedad y a excluir algunos casos con indicación de tratamiento. Sin embargo, el empleo de RM en el seguimiento periódico de la respuesta a la TES es prácticamente general. No hay que olvidar, en cualquier caso, que la RM debería emplearse en el 100% de los casos en edad adulta, especialmente siendo la disponibilidad de esta técnica tan generalizada.
Otro aspecto sorprendente, a la vez que preocupante, es la infrautilización de la determinación de quitotriosidasa (QT) plasmática. Esta enzima de origen macrofágico, se encuentra muy elevada –de un modo casi exclusivo– entre los pacientes con EG (13), además sus valores plasmáticos reflejan la carga total de células de Gaucher del organismo (14) y su variación tras el inicio de la TSE permite monitorizar la respuesta al tratamiento (15,16). En todas las guías al uso la determinación periódica de QT se considera obligada y por este motivo desde la FEETEG y el Grupo Español de trabajo en EG se implementó un sistema gratuito de cobertura nacional para la determinación de este marcador entre los pacientes con EG. A pesar de ello tan solo un 60% de los casos referidos tienen determinaciones seriadas de QT. Esta es una cifra excesivamente baja, especialmente considerando su alto valor informativo y el coste nulo para los centros.
En conclusión, puede decirse que la red sanitaria pública, que soporta un alto coste en el tratamiento de la EG, está bien dotada para ello, tanto en los aspectos diagnósticos, como terapéuticos, en los dos niveles analizados, el de los hospitales de referencia y los comarcales. La eficiencia en el empleo de recursos tecnológicos en los pacientes con EG es, aunque aceptable para una enfermedad podo frecuente y dispersa geográficamente, inferior a lo deseable. Por ello, debería hacerse un esfuerzo adicional para corregirlo, incidiendo muy especialmente en la necesidad de generalizar del empleo de la RM en la evaluación inicial, especialmente, y el seguimiento, así como de la determinación periódica de QT en todos los pacientes con EG.
A pesar de la creciente sensibilidad por las denominadas “enfermedades raras” en general y en la de Gaucher en particular, estas siguen siendo rarezas clínicas aisladas en un marco de alta presión asistencial. El esquema asistencial desarrollado en torno a la EG en España está demostrando su valor y puede, con gran probabilidad, servir de modelo para otras enfermedades metabólicas que ya pueden ser tratadas de modo similar (17) o lo serán en un futuro inmediato. Cualquier esfuerzo encaminado a mejorar su grado de conocimiento y control redundará, plausiblemente, en beneficios a la hora de administrar los recursos terapéuticos con eficacia y eficiencia.
AGRADECIMIENTOS
Este estudio ha sido subvencionado mediante una ayuda de la Fundación Española para el Estudio y Terapéutica de la Enfermedad de Gaucher.
Los autores desean agradecer la colaboración expresa de los siguientes médicos pertenecientes al “Grupo Español de Trabajo en Enfermedad de Gaucher”.
José Albadalejo Méndez (Hospital Ntra. Sra. del Rosel, Murcia); Koldo Atutxa Aresti (Hospital Galdakao, Bizkaia); J. Calabuig (Hospital La Fe, Valencia); Gregorio Castellano Tortajada (Hospital 12 de Octubre, Madrid); J. Dalmau (Hospital Infantil La Fe, Valencia); Javier de la Serna (Hospital 12 de Octubre, Madrid); José L. Fernández Epifanio (Hospital Virgen del Puerto, Plasencia); Mª Angeles Fernández Galán (Hospital Virgen del Puerto, Plasencia); Joaquín Fernández Garcia (Hospital de Cabueñes, Gijón); J. Gómez (Hospital Infantil La Fe, Valencia); Miguel Gracia Antequera (Hospital Dr. Peset, Valencia); Angela Ibáñez García (Hospital General, Albacete); Roberto López González (Hospital del Insalud, León); Juan López Muñoz (Hospital Torrecárdenas, Almería); Guillermo Martín Núñez (Hospital Virgen del Puerto, Plasencia); Pedro Martínez Odriozola (Hospital de Basurto, Bilbao); J. Martínez Pardo (Hospital Ramón y Cajal, Madrid); Manuel Muñoz Rodríguez (Hospital del Insalud, León); José A. Nieto Rodríguez (Hospital Virgen de la Luz, Cuenca); Mª Angeles Ruiz (Hospital Francesc Borja, Valencia); A. Vallcanera (Hospital Infantil La Fe, Valencia).
Bibliografía
1. Devine EA, Smith M, Arredondo Vega FX, Shafit Zagardo B, Desnick RJ. Chromosomal localization of the gene for Gaucher disease. Prog Clin Biol Res 1982; 95: 511-534. [ Links ]
2. Horowitz M, Wilder S, Horowitz Z, Reiner O, Gelbart T, Beutler E. The human glucocerebrosidase gene and pseudogene: structure and evolution. Genomics 1989; 4: 87-96. [ Links ]
3. Grabowski GA. Gaucher disease. Enzymology, genetics, and treatment. Adv Hum Gen 1993; 21: 377-441. [ Links ]
4. Giraldo P, Pérez Calvo JI, Giralt M. Características clínicas de la enfermedad de Gaucher en España. Resultados preliminares de una encuesta nacional. Med Clín (Barc) 1997; 109: 619-622. [ Links ]
5. Zimran A, Kay A, Gelbart T, Garver P, Thurston D, Saven A, Beutler E. Gaucher Disease: Clinical, Laboratory, Radiologic, and Genetic features of 53 patients. Medicine (Balt) 1992; 71: 337-353. [ Links ]
6. Meeker DP. Pulmonary manifestations of Gaucher disease. Gaucher Clin Pers 1996; 4: 1-5. [ Links ]
7. Autor corporativo. ICCGD Registry Update. Skeletal disease among patients in the ICGG Registry. October, 1994. [ Links ]
8. Pastores GM, Einhorn T. Skeletal complications of Gaucher disease: pathophysiology, evaluation, and treatment. Sem Hematol 1995; 32 (Supl. 1): 20-27. [ Links ]
9. Guías de actuación en pacientes adultos con enfermedad de Gaucher tipo 1. Fundación Española para el Estudio y Terapéutica de la Enfermedad de Gaucher. Zaragoza, 2002 [ Links ]
10. Roca M, Mota J, Giraldo P, Pérez Calvo JI, Gómez Pereda R, Giralt M. La resonancia magnética en el diagnóstico de extensión y de las complicaciones de la enfermedad de Gaucher tipo I. Rev Clin Esp 1997; 197: 550-554. [ Links ]
11. Duursma SA, Aerts JMFG, Belmatoug N, Cox TM, Einhorn T, Esplín JA, Hollak CEM, et al. Management of Gaucher disease. Current status and future directions: a roundtable discussion. Sem Hematol 1995; 32 (Supl. 1): 45-52. [ Links ]
12. Pérez Calvo JI, Pastores GM. Tratamiento de la enfermedad de Gaucher tipo I. ¿Hacia la solución definitiva? Med Clín (Barc) 1994; 103: 16-18. [ Links ]
13. Hollak CE, van Weely S, van Oers MH, Aerts JM. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J Clin Invest 1994; 93 (3): 1288-92. [ Links ]
14. Hollak CE, Aerts JM. Clinically relevant therapeutic endpoints in type I Gaucher disease. J Inherit Metab Dis 2001; 24 (Supl. 2): 97-105. [ Links ]
15. Pérez Calvo JI, Giraldo P. Tratamiento de la enfermedad de Gaucher tipo 1. Mucho camino todavía por recorrer. Med Clín (Barc) 2000; 114: 774-776. [ Links ]
16. Giraldo P, Cenarro A, Alfonso P, Pérez-Calvo JI, Rubio-Félix D, Giralt M, Pocoví M. Chitotriosidase genotype and plasma activity in patients type 1 Gaucher's disease and their relatives (carriers and non carriers). Haematologica 2001; 86: 977-984. [ Links ]
17. Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, et al. Safety and efficacy of recombinant human a-galactosidase A replacement therapy in Fabry's disease. N Engl J Med 2001; 345: 9-16. [ Links ]

