










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versão impressa ISSN 1130-0108
Rev. esp. enferm. dig. vol.104 no.11 Madrid Dez. 2012
https://dx.doi.org/10.4321/S1130-01082012001100007
Influence of genetics on tumoral pathologies: The example of the adenocarcinoma arising in Barrett's esophagus
Influencia de la genética en la patología tumoral. El ejemplo del adenocarcinoma desarrollado sobre el esófago de Barrett
Vincenzo Villanacci1, Gabrio Bassotti2, Marianna Salemme1 and Elisa Rossi3
1 Department of Pathology. Spedali Civili. Brescia, Italy.
2 Gastroenterology and Hepatology Section. Department of Clinical and Experimental Medicine. University of Perugia. Italy.
3 Centro de Investigaciones Biológicas. Consejo Superior de Investigaciones Científicas (CSIC), and Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER). Madrid, Spain
ER is supported by the JAE-Doc Program of CSIC funded by FEDER.
ABSTRACT
Barrett's esophagus (BE) refers to an abnormal change (metaplasia) in the cells of the inferior portion of the esophagus. About 10% of patients with symptomatic gastroesophageal reflux disease (GERD) have BE. In some cases, BE develops as an advanced stage of erosive esophagitis. The risk of esophageal cancer appears to be increased in patients with BE. The only way to diagnose BE is by endoscopy and histology. Some studies suggest that intensive treatment of Barrett's esophagus with effective acid suppression can reduce the amount of abnormal lining in the esophagus. It is not clear whether such treatment also prevents esophageal cancer. Generally, the cancer starts out as carcinoma of the esophagus on the surface, and then invades the surrounding tissue. Surgery offers the best chance of long-term survival. There are many events that occur in Barrett's esophagus that lead to the development of cancer and most of them appear to occur early, before high-grade dysplasia or cancer develops. No one knows what the late events are and how cells acquire the ability to leave their normal growth boundaries. It is now widely accepted that the development of most cancers is due to something called genomic or genetic instability. The aim of this review is to show BE pathology in its progression to cancer looking for new biomarkers to distinguish between BE-dysplasia (low grade and high grade)- adenocarcinoma (ADC) and to characterize the ADC, giving more hope for its treatment.
Key words: Adenocarcinoma. Barrett's esophagus. Dysplasia. Molecular markers.
Introduction
Barrett's esophagus - A pathological view
The pioneer thoracic surgeon Norman Barrett (1903-1979) is chiefly remembered for his description of the esophagus lined by columnar epithelium, a condition subsequently referred to as Barrett's esophagus (BE). Barrett's epithelium is now the subject of intensive clinical and biological study. It is the most important known risk factor for adenocarcinomas of the esophagus and gastric cardia (sometimes termed Barrett's carcinomas or adenocarcinomas). These cancers are increasing in incidence at a dramatic rate in many Western societies, especially among white men (1). The 2008 updated guidelines for the diagnosis, surveillance and therapy of BE endorsed by the American College of Gastroenterology (ACG) defines BE as "a change in the distal esophageal epithelium of any length that can be recognized as columnar type mucosa at endoscopy and confirmed to have intestinal metaplasia by biopsy of the tubular esophagus" (2). The definition of BE varies worldwide, particularly with regard to the need to identify goblet cells in esophageal biopsies in order to diagnose this condition (Fig. 1). Most authors, including us, consider intestinal metaplasia as the epithelial type that facilitates cancer development. In literature it has been demonstrated that a Barrett adenocarcinoma may also arise in a cardiac-type background mucosa, without goblet cells. In fact, in the series reported by Takubo et al. (3) more than 70% of small primary adenocarcinomas of the esophagus were located adjacent to cardiac/fundic-type rather than to intestinal-type mucosa. This point of view is also reported by Riddel and Odze (4).
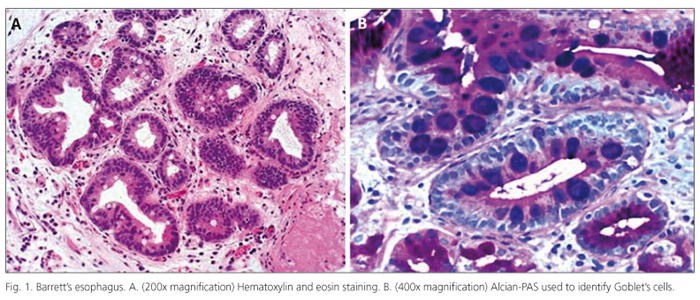
At present, it is unclear to what extent the goblet and non-goblet cell population in patients with BE are related. Most authorities define "intestinal metaplasia" by the presence of goblet cells even though absorptive cells, endocrine cells, and Paneth cells may all be present in patient with BE. However, emerging evidence suggests that the background non-goblet columnar epithelium in BE may, in fact, already be "intestinalized" (5) by revealing positivity for a variety of peptides, such as CDX2, Hep Par 1, Villin and DAS-1, which represent proteins and transcription factors specific for intestinal differentiation in the normal gastrointestinal tract.
Problems related to the need to identify goblet cells to diagnose BE include the facts that goblet cells are uncommon in pediatric patients with BE and that the sampling error is a major limitation to the diagnosis of BE in endoscopically obtained mucosal biopsies.
There are many events or steps that occur in BE that lead to the development of cancer. A few of these events are known but most are not. Most of the known events appear to occur early, before high-grade dysplasia or cancer actually develops.
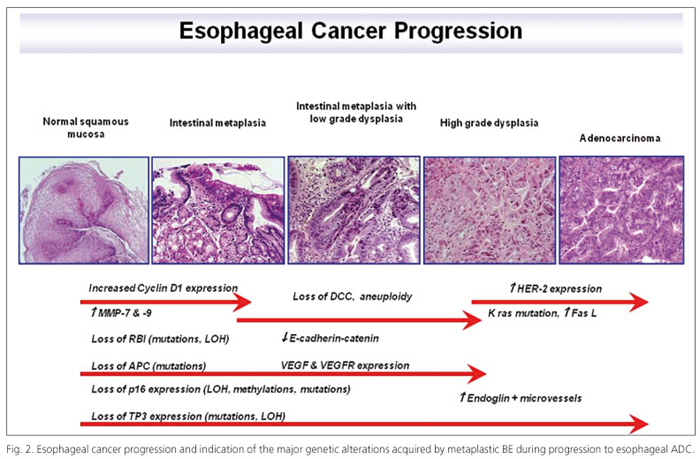
No one knows what the late events are that give cells the ability to leave their normal growth boundaries and become a cancer. It is now widely accepted that the development of most cancers is due to something called genomic or genetic instability. This theory was first proposed by Dr. Peter Nowell in 1976. The theory is that for some unknown reason, perhaps due to environmental factors or inherited factors, some cells in the body develop genetic abnormalities that give them the ability to outgrow genetically normal cells. These abnormal cells grow and expand into a clone of cells (a group of cells having the same genetic make-up) and may replace their neighboring normal cells. Eventually one of the abnormal clones may undergo another genetic change that leads to the development of a sub-clonal population with the expansion of this cell line into its own large clone of cells. As multiple genetic abnormalities occur, multiple sub-clones develop or evolve. Eventually, one of these sub-clones may acquire the necessary combination of genetic abnormalities to become a cancer. Cancer in BE develops in a linear fashion from metaplasia to dysplasia to cancer. This progression occurs through clonal evolution similar to that proposed by Nowell, but has been shown to be more complex, with multiple subclones developing in the Barrett's tissue prior to the development of cancer. Flow cytometric abnormalities can be detected early in BE and before the development of high-grade dysplasia and cancer. These abnormalities include increased 4N and aneuploid cell populations. Genetic abnormalities in the p53 and p16 genes with loss of function in these genes, occur even earlier than flow cytometric abnormalities. Previous research (Fig. 2) showed that p53 gene and p16 gene abnormalities are present in the vast majority of patients with a Barrett's associated cancer. P53 gene abnormality greatly increases the risk of developing cancer in BE, while p16 gene abnormalities are the earliest gene abnormalities so far detected in BE, present in more than 85% of Barrett's linings. It is hypothesized that p16 abnormalities contribute to the expansion of Barrett's cells along the surface of the esophagus, as well as to the expansion or spread of additional gene abnormalities that occur during progression to cancer in BE. Other genes develop abnormalities in the progression to BE but their relationship to flow cytometric abnormalities or the development of cancer is less clear than those of p53 and p16. Identification of additional genes will lead to a better understanding of how cancer develops, tests to determine who is at risk for developing cancer, and better therapy in the treatment of cancer and BE.
From metaplasia to adenocarcinoma
It is well known that metaplasia is a conversion of one cell type to another (6) and it seems that metaplasia predisposes to the development of dysplasia and subsequently neoplasia. For this reason understanding the steps leading to cancer and studying prognostic and diagnostic markers for tumor progression provide novel target for gene therapy. Nowadays a lot of techniques help us in studying the cancer development such as comparative genomic hybridization (CGH), fluorescence in situ hybridization (FISH), polymerase chain reaction (PCR), immunohistochemistry (IHC); but some of the last methodologies (gene microarrays, gene expression profiling) allowed us to identify more genes specifically involved in this process (7). The cause of metaplasia is presumably a chance in expression of genes whose normal function is to distinguish the two tissue types in normal development (6). P63, a member of the p53 family of transcription factors, seems to have a role in the normal development of the esophageal epithelium suggesting it could be one of the master switch genes. Barrett's metaplasia shows multiple genetic alterations. Only about 10% of individuals with reflux develop BE, indicating that additional factors are involved in this progression. A comparison of individuals with gastroesophageal reflux disease (GERD) who did and did not progress to BE revealed that those who developed BE had a characteristic profile of risk factors. The main one of these was bile reflux, but hiatal hernia, defective lower esophageal sphincter pressure, and longer episodes of reflux were the other key determinants (8). Presumably, not a single gene might be able to discriminate those patients that will progress to high grade dysplasia (HGD)/adenocarcinoma (ADC). Some investigations (9) show that the selection of markers based on DNA array experiments may provide molecular criteria for discrimination of pathologic conditions of esophageal epithelium and that the expression analysis of a limited number of highly selected genes may have clinical usefulness for the treatment of patients with this disease. The genes involved in the development of Barrett's metaplasia seem to be: p16, APC, Rb, p53, DCC which are lost in this early step. Moreover, cyclin D1 could be amplified, p53 mutated, APC hypermethylated and Bcl2, iNOS, COX-2, CDX2, SRC, SKI, SnoN overexpressed (9-11).
Aneuploidy (abnormal chromosome numbers) has been strongly associated in disease progression and predicts ADC development (12). Aneuploidy has been detected in ADC, dysplastic tissue and metaplastic Barrett's epithelium (13) but others authors suggested that aneuploidy characterizes more the dysplastic and the tumor development then the metaplastic step (14). Cell signalling genes such as HER-2 (also called ERBB2, HER-2/neu, NEU), EGFR, TGFα gene, Kras seems to be overexpressed/amplified in adenocarcinomas arisen in BE but not in metaplasia, appearing to be a late event (9,15). Conversely, TFF1, MUC5AC, meprin A and sucrose isomaltase are always positive in non dysplastic BE and negative in dysplasia (16). Recent data suggest that the stem cells of gastrointestinal tumors may express the same stem cell markers as the normal intestinal epithelium. At the moment, no markers of stem cells in metaplastic BE has been identified. However, since the epithelium of BE is a form of incomplete intestinal metaplasia, perhaps Lgr5 and DCAMKL-1 may be use in the search for stem cells in BE and esophageal ADC (17). Development of ADC in BE follows a characteristic metaplasia-dysplasia adenocarcinoma sequence.
Two patterns of Barrett dysplasia have been recently described (18): adenomatous (type I) and non-adenomatous (type II or foveolar type). The former is said to account for the majority of cases, while the latter is uncommon and it has been less characterized.
Adenomatous-type dysplasia is composed of glands or villous structures lined by tall columnar cells with hyperchromatic nuclei and dense eosinophilic cytoplasm; goblet cells are often identified. Foveolar type dysplasia is characterized by cuboidal to columnar cells with pale clear to light eosinophilic cytoplasm and round to oval nuclei; goblet cells are absent. Regarding immuphenotype, MUC2, CDX2 and villin are markers of intestinal differentiation and so are useful to diagnose type I dysplasia; by contrast, foveolar type dysplasia commonly expresses MUC5AC. Brown et al. (18) placed emphasis on these different pathological entities, highlighting the importance of the morphological subclassification of BE dysplasia into adenomatous and gastric foveolar types. In particular, the foveolar dysplasia should be taken into consideration, as part of a non-intestinal neoplastic pathway.
Dysplasia is classified in low and high grade dysplasia (LGD and HGD), depending on the severity of atypical cytological alterations and nuclear polymorphism. LGD means that there are some atypical changes but these changes do not involve most of the cells, and the growth pattern of the glands is still normal. In LGD some of the nuclei, less than 50%, are large and have dark spots but the cells are still growing in an even row. Some cells are dividing (a process called mitosis which usually indicates increased growth rate), but very few. HGD is considered the most advanced dysplasia with atypical changes in many of the cells and a very abnormal growth pattern of the glands. In HGD, the growth pattern of the glands, or rows of cells, are distorted or very irregular. Some of the glands are branching or budding. More than 50% of the cells have large spotted nuclei and are frequently dividing. The number of alcian blue staining goblet cells is reduced. The cellular cytoplasm is reduced and looks abnormal. It is well known gastrointestinal reflux is the principal trigger for this sequence but the histological assessment of dysplasia and prediction of cancer risk is subjective and depends on inter-observer variability (19). The progression from metaplasia through dysplasia to cancer appears to be a multi-step process with accumulation of somatic mutations. The development of aneuploidy seems to play an important role (14,20) and some authors suggested that abnormal DNA ploidy status is a prognostic factor for BE progression into ADC (21). Changes in cellular DNA content and expression levels of p53 and Ki67 in BE are associated with the development ADC and might serve as markers to identify ADC at an early stage (22). E-cadherin decreased expression and APC gene inactivation also seems to be involved in dysplasia steps and evolution (23). Moreover, some authors proved that β-catenin can be helpful for a diagnosis of LGD in BE, although it stains positively in a subset only, whereas p53 remains an appropriate marker to define HGD. In case of doubt, cyclin D1 can be added to separate LGD from HGD in BE (24) while Scheil-Bertram and colleagues (25) investigated alpha-methylacyl coenzyme A racemase (AMACR) proving this might be a new diagnostic marker for dysplasia-carcinoma sequence in Barrett's low-grade neoplastic lesions. In addition, topoisomerase II (TOPOII), S100A9 and lipocalin-2 resulted up-regulated and overexpressed by microarray analysis, RT-PCR and IHC (16) in ADC. In some studies it was proved that the amplification/overexpression of HER-2 characterizes the presence of dysplasia (15). Finally, dysplasia is the most predictive marker for risk of esophageal ADC, whereas endoscopic and histological diagnoses are still the gold standard for surveillance of patients with BE. However, both are limited, either by sampling errors in biopsies or by differences in histological interpretation. Several studies try to identify candidate biomarkers that may have predictive value and may serve as additional factors for the risk assessment of esophageal ADC. The major risk factors for esophageal ADC are GERD and BE because they predispose to malignancy (Fig. 2).
Environmental risk factors for esophageal adenocarcinoma: GERD, obesity, Helicobacter pylori (Hp), alcohol and tobacco
GERD and its sequel BE are the major risk factors for esophageal ADC (26,27). GERD is thought to be the factor that both injures the esophageal squamous epithelium and provides the abnormal background necessary to healing the reflux esophagitis through metaplasia rather than through the regeneration of squamous epithelium. The specialized intestinal metaplasia of BE appears to be more resistant to acid-peptic damage than the native squamous epithelium, but for reasons that are not clear, is predisposed to carcinogenesis. Indeed, the large majority of esophageal adenocarcinomas appear to arise from this specialized intestinal metaplasia (27). Obesity has been established as a strong risk factor for esophageal ADC. A recent systematic review of the literature found a positive association between body mass index (BMI) and the risk of esophageal ADC (28). Other recent data has found a stronger association of esophageal ADC with central (abdominal) obesity than BMI alone and a strong association between central obesity and BE have also been reported (29). Central obesity may predispose towards GERD by increasing pressure within the abdomen. In addition, obesity may alter circulating levels of pro-proliferative factors so as to promote esophageal carcinogenesis. Hp has been classified by the World Health Organization's International Agency for Research on Cancer as a group I carcinogen for ADC of the distal stomach. In contrast, it seems that there is a significant inverse relationship between esophageal ADC and Hp infection because Hp infection decreases gastric acid secretion and consequently prevents the development of GERD (30). Unlike esophageal squamous cell cancer where alcohol and tobacco use are strong risk factors, a number of studies have found that there is only a moderate association of tobacco use with esophageal ADC and no clear association with alcohol use.
Molecular pathogenesis of esophageal ADC arising in BE
Metaplasia (the process in which one adult cell type replaces another) is one way in which tissues respond to chronic inflammation. Although the metaplastic cells may be more resistant to the inflammatory insult than the native cells, the metaplasia may also predispose towards malignancy, in other words BE predisposes towards developing ADC by the following steps:
Proliferation without exogenous stimulation
In general, it is the expression of oncogenes that allows cells to proliferate without exogenous stimulation. Proto-oncogenes are normal cellular genes that promote cell growth. Oncogenes are proto-oncogenes that have become overactive as the result of mutation. Examples of oncogenes implicated in the development of esophageal ADC are cyclins D1, E, B1 and A. Cyclins D1 and E, along with their cyclin-dependent kinases (cdks), regulate the pivotal G1 to S transition point in the cell cycle. Cyclin A is expressed during the S and G2 phases, whereas cyclin B1 acts to control the G2 to M transition. Increased nuclear expression of cyclin D1 protein has been detected in biopsy specimens of non-dysplastic Barrett metaplasia, suggesting that it may play an early role in carcinogenesis (31). In contrast, overexpression of cyclin E has been found in dysplastic Barrett epithelium and in ADC, but not in non-dysplatic BE (32), while expression of cyclin B1 has been detected in nondysplatic and dysplatic BE as well in Barrett ADC. Cyclin A expression has been found to increase as the metaplasia progresses through dysplasia to ADC (33,34). In addition to the direct activation of oncogenes, alterations in growth factors, growth factor receptors, or the signaling pathways that mediate growth factor-receptor interactions, can also allow cells to proliferate without exogenous stimulation. For example, increased expression of epidermal growth factor (EGF), transforming growth factor-α (TGF-α) and EGF receptor (EGFR or HER-1) have been found in esophageal adenocarcinomas. Increased expression of TGF-α and the EGFR have been found to occur early in non-dysplastic Barrett epithelium (35,36). The role of the oncogenic form of the normal EGFR family member HER-2 in esophageal ADC progression remains controversial. However recent data suggest that HER-2 amplification may be associated with a worse outcome for esophageal ADC. Downstream of tyrosine kinase receptors like EGFR are the Ras proteins (including H-ras and K-ras), which play a central role in the regulation of cell proliferation. K-ras mutations have been reported in 11-40% of esophageal adenocarcinomas (37).
Resistence to growth-inhibitory signals
Tumor suppressor genes are normal genes that usually function to restrain cell growth. Cells can acquire the ability to resist growth inhibitory signals by inactivating tumor suppressor genes through one or a combination of three mechanisms including mutation of the gene, loss of heterozygosity (LOH, which is a deletion of the chromosomal region containing the gene), or promoter methylation (attachment of methyl groups to the promoter region of genes). Tumor suppressor genes implicated in the progression of Barrett metaplasia to cancer have shown inactivation by all of these mechanisms. Examples of tumor suppressor genes implicated in the development of esophageal ADC include p16, p53, p14ARF, p27, and the APC gene. P16 and p53 proteins normally act to block cell cycle progression at the G1 to S transition and, therefore, inactivation of the p16 or p53 genes enables unregulated cell growth. Allelic loss of 9p21, the chromosomal locus for p16, and methylation of the p16 promoter have been reported in 45-54% of esophageal adenocarcinomas (38). Moreover, p16 mutation, LOH or promoter methylation has been detected in non-dysplastic Barrett's metaplasia in approximately 80% of cases, suggesting that genetic alterations of p16 are among the earliest events in the neoplastic progression of BE (39).
Avoidance of apoptosis
Normal cells have the ability to destroy themselves through the process of apoptosis, a genetically regulated, innate form of cell suicide. This process prevents the survival of cells that have sustained cancer-promoting injuries that might threaten the organism. The cellular apoptotic machinery can be triggered by a number of factors including DNA damage, death receptor activation, and metabolic abnormalities. Once activated, the apoptotic machinery leads to cell death through activation of an executioner pathway (40). Tumor cells must find ways to avoid apoptosis if they are to survive. In addition to its tumor suppressor activity p53 protein also functions as an initiator of apoptosis. Esophageal ADC cell can avoid apoptosis by inactivating p53. Apoptosis also can be initiated when death receptors on the cell surface bind with ligands such as Fas-ligand (FasL) and TNF-related apoptosis inducing ligand (TRAIL). The Fas death receptor is normally found on gut epithelial cells, whereas lymphocytes express both Fas death receptor and FasL, which can bind the Fas receptor on the surface of tumor-killing lymphocytes, thereby destroying the lymphocytes that might attack the cancer cells.
Recently, expression of TRAIL has been found to decrease progressively as metaplastic Barrett epithelium develops dysplasia and carcinoma (41). Synthesis of an agent that blocks apoptosis is another mechanism whereby cancer cells avoid their own destruction. For example, esophageal adenocarcinomas can exhibit increased expression of cyclooxygenase-2 (COX-2), which has been shown to decrease apoptosis rates in esophageal ADC cells in vitro (42). COX-2 overexpression also has been detected in benign Barrett metaplasia, and COX-2 expression has been found to increase as the cells progress to dysplasia and carcinoma (43).
Resistance to cell senescence
Senescence, like apoptosis, is an innate mechanism that limits the proliferation in normal cells. As cells undergo successive divisions, their telomeres, which are short repetitive DNA sequences located at the ends of chromosomes, undergo progressive shortening. Once the telomeres shorten to a critical length, the cell enters senescence, a permanent state of growth arrest. Therefore, for cells to replicate indefinitely (i.e. to become immortal), telomere length must be maintained. Telomerase is the enzyme responsible for the synthesis and maintenance of telomeres. High levels of telomerase expression have been found in esophageal ADC, whereas low expression levels are found in non-dysplastic Barrett epithelium. Moreover, a marked increase in telomerase expression occurred during the transition from LGD to HGD in Barrett epithelium (44).
Development of new vascular supplies (angiogenesis)
For tumors to grow, they must form new blood vessels to provide nutrients and eliminate metabolic waste products. Vascular and endothelial growth factors (VEGFs) are potent promoters of angiogenesis. VEGF expression has been found to be significantly increased in esophageal adenocarcinomas compared to dysplastic and metaplastic BE and normal esophageal mucosa. Endoglin (CD105), a homodimeric cell-surface glycoprotein component of the transforming growth factor β (TGF-β) receptor complex, has been identified as a proliferation-associated marker of endothelial cells of tissue undergoing neovascularization, such as regenerating and inflamed tissues (45). Furthermore, it was suggested that CD105 expression correlates closely with cell proliferation markers in tumor endothelial cells (46). In esophageal ADC, the number of tumor microvessels that are positive for endoglin has been found to correlate significantly with angiolymphatic invasion, lymph node metastasis, and overall prognosis. Moreover, Barrett epithelium with HGD contains a significantly greater number of endoglin-positive microvessels than BE with LGD (47).
Invasion and metastasis
To invade and metastasize, tumor cells must lose their cell-cell adhesion and acquire the ability to degrade the extracellular matrix. Cadherins are a large family of adhesion molecules that are located on the cell surface, where they bind to cadherins on the surface of neighboring cells. The cadherins are anchored in place by binding to catenins, which are attached to the cell cytoskeleton. Loss of cell-cell adhesion by failure of cadherins to interact with either the catenins or with other cadherins can predisposes towards invasion and metastasis. As the degree of dysplasia in Barrett epithelium increases, there is a decrease in membranous E-cadherin and β-catenin and an increase in the cytoplasmic and nuclear location of these proteins (47). Matrix metalloproteinases (MMPs) are a family of proteolytic enzymes that mediate the destruction of the extracellular matrix, allowing for tumor invasion and spread. MMPs-7 and -9 have been found to be increased in non-dysplastic BE, with even higher levels found in dysplastic Barrett mucosa and esophageal ADC (48).
Treatment of GERD and Barrett's adenocarcinoma
Esophageal acid exposure is important in the pathogenesis of BE, and possibly in the progression of BE to dysplasia and carcinoma. Some studies suggests that PPI therapy is associated with a significant reduction in the risk of developing dysplasia in patients with BE (49), however many cases develop ADC in BE. For this reason there are guidelines for the diagnosis and treatment of gastroesophageal reflux disease. Diagnostic guidelines in GERD address empiric therapy and the use of endoscopy and ambulatory reflux monitoring; esophageal manometry is not mandatory in respect to endoscopy (50). Treatment guidelines address the role of lifestyle changes, patient directed (OTC) therapy, acid suppression, promotility therapy, maintenance therapy, antireflux surgery, and endoscopic therapy in GERD (51). Local endoscopic treatment represents an alternative to esophageal resection in the case of intraepithelial high-grade dysplasia and selected early adenocarcinomas in BE. When an ADC appears, the only possibility is surgery, while radiation therapy offers tumor control, however it is more effective on small tumors; and sometimes chemotherapy is added to radiation therapy. If a tumor is blocking the esophagus, laser therapy, photodynamic therapy or stenting may be used to create an opening so that swallowing is easier.
Emerging markers
HER-2 oncogene, TOPOIIα, GATA6
HER-2, a gene located on chromosome 17q11.2-12 and activated by amplification, encodes the epidermal growth factor receptor 2 (HER-2), which belongs to the human epidermal growth factor receptor family. HER-2 is a protooncogene, frequently amplified/overexpressed in breast cancer and in other carcinomas such as adenocarcinomas arising in BE (52,53).
It has been shown that HER-2 amplification/overexpression has therapeutic and prognostic implications in breast cancer and other carcinomas (52,54,55), but the association of HER-2 alterations with the histologically proposed metaplasia-dysplasia-adenocarcinoma sequence is controversial. Concerning BE, some studies suggested that HER-2 overexpression is a frequent and early event (35) whereas in others this occurrence was much less common, appearing relatively late in BE tumorigenesis (56). In particular, HER-2 was negative in all BE cases and positive in 55% of cases with dysplasia and in 24% of cases with associated ADC; other authors, applying locus-specific probe, demonstrated HER-2 gene amplification in 19% or in the 35% of BE with ADC (57). From a clinical point of view, patients with HER-2-positive tumors, such as ADC arising in BE, had a significantly poor prognosis compared with patients with negative tumors. Therefore, HER-2 (gene/protein) might be used as a prognostic index in patients with these neoplasms. This seems particularly interesting, because an antibody-based therapeutic approach (trastuzumab) targeting this protein has been reported as an effective adjunctive treatment for breast neoplasms overexpressing HER-2 (57). HER-2 status can be measured by several methods, targeting the protein by immunohistochemistry, Western blotting, enzyme-linked immunosorbent assay, or the mRNA by reverse transcriptase-polymerase chain reaction and Northern blotting or targeting the DNA by fluorescence in situ hybridization (FISH), chromogenic in situ hybridization, and Southern blotting. There is no consensus with regard to the optimal test for HER-2 assessment. This lack of consensus may in part be logistical, and not entirely based on the available science and clinical data.
The most widely used assays are immunohistochemical analysis and FISH, both Food and Drug Administration (FDA) approved, which measure protein expression and gene amplification respectively. In breast cancer a treatment with trastuzumab is generally suggested when a concordance between these two techniques is found (58). Immunohistochemical studies reported 11 to 73% variation of HER-2 overexpression in invasive ADC associated with BE, possibly reflecting the well-known inherent problems of immunohistochemistry in quantification and interpretation of the HER-2 status (59).
We reported in a previous paper (60) the potential critical role for HER-2 in the pathogenesis of esophageal carcinoma (Fig. 3). Although this study reports on a small series and validation is required on a larger analysis, it seems that several points merit attention: a) HER-2 is associated with distinct stages of dysplasia arising in BE; and b) HER-2 is associated with the progression of dysplasia and shows a trend toward a shorter time-to-progression (TTP). The end-points were the occurrence of progression and the TTP from the initial histologic lesion to the worst pathological pattern. So, according with other articles which studied the risk of BE progression by endoscopic and histological features (61), we think that the identification of biomarkers could help in the evaluation of cancer TTP.
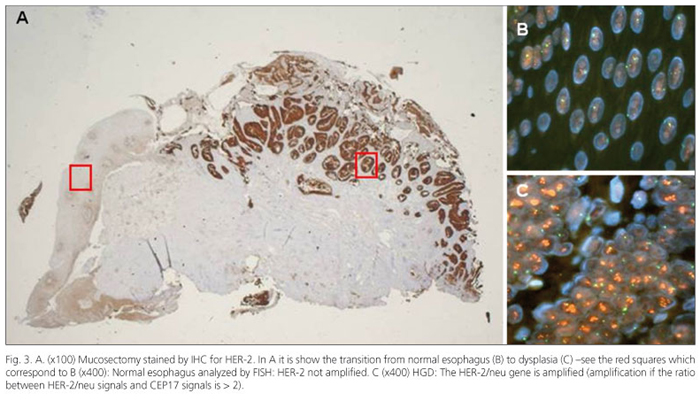
Recently, we considered HER-1(EGFR), another member of the epidermal growth factor family (EGF-family). Both EGFR and HER-2 receptors are targets for immunotherapy, and for this reason their protein expression and gene amplification are widely investigated. In this case, studying EGFR we found a different behavior compared to HER-2, in fact IHC proved that EGFR protein overexpression in gastrointestinal cancers is common but FISH assessment showed that EGFR gene amplification is rare (62).
TOPOIIα gene as well HER-2 is located on chromosome 17q, and they can be co-amplified in cancer. Amplification of both genes has been reported in breast, prostate, gastric, colorectal, pancreatic and esophageal carcinomas (63,64). There are 2 isoforms of mammalian topoisomerase II, α and β (65). DNA topoisomerase II catalyzes a transient double strand DNA break, which allows the passage of another DNA duplex through the break before the strands are resealed. TOPOIIα represents the target enzyme for specific anticancer drugs, such as anthracyclines, commonly used for a variety of both hematological and solid cancers, including leukemias, lymphomas and breast cancer. In vitro studies have shown a correlation between the expression level of TOPOIIα in cancer cells and the sensitivity of those cells to topoisomerase inhibitors (66).
Gene amplification is a tumor-specific event during malignant transformation. Recent studies have proposed a lineage-dependency (addiction) model of human cancer whereby amplification of certain lineage transcription factors predisposes a survival mechanism in tumor cells. It was demonstrated that recurrent amplification at 18q11.2 occurs in 21% of esophageal adenocarcinomas. Using an integrative genomic strategy reveals a single gene, the embryonic endoderm transcription factor GATA6, as the selected target of the amplification. Overexpression of GATA6 is found in esophageal adenocarcinomas that contain gene amplification. Patients whose tumors carry GATA6 amplification have a poorer chance of survival. We show that ectopic expression of GATA6, together with FGFR2 isoform IIIb, increases anchorage-independent growth in immortalized Barrett's esophageal cells. Conversely, siRNA-mediated silencing of GATA6 significantly reduces both cell proliferation and anchorage-independent growth in esophageal ADC cells. It seems that selective gene amplification of GATA6 during cancer development sustains oncogenic lineage-survival of esophageal ADC (67).
COX-2, CDX2, CDC-2
Cycloxygenase 2 (COX-2) is a protein which acts as an enzyme and specifically catalyzes the production of certain chemical messengers called prostaglandins. Some of these messengers are responsible for promoting inflammation. When COX-2 activity is blocked, inflammation is reduced. An increased expression of COX-2 was found in both BE and ADC (68), therefore the use of COX-2 inhibitors might have a role to reduce progression, but studies are limited (69). Caudal-type homeobox transcription factor 2 (CDX2) is the gene that directs early embryogenesis in mice. It is required to form the placenta. Ectopic expression of CDX2 was reported more than 85% of the human patients with acute myeloid leukemia (AML). Ectopic expression of CDX2 in murine bone marrow induced AML in mice and upregulate Hox genes in bone marrow progenitors (70). In other studies CDX2 seems to be important because its expression appears to be largely restricted to cells of intestinal derivation and it is thought to have an important role in the early differentiation and maintenance of intestinal epithelium through the transcription of an intestinal specific gene. Thus, CDX2 may be helpful for a more precise diagnosis of BE (71).
Cell division cycle 2/cyclin-dependent kinase 1 (CDC2/CDK1) is a catalytic subunit of a protein-kinase complex which induces the onset of mitosis and it is universally present in eukaryotes. Only a few studies exist on the immunohistochemical expression of CDC2 in esophageal ADC and in BE in particular (72). Some COX-2 studies in this setting did not find differences between BE with and without dysplasia (73,74), whereas other authors reported an increase of COX-2 expression by immunohistochemistry in the sequence BE-LGD-HGD-ADC, confirmed by immunoblotting (43), and a significant increase in the progression from LGD to HGD was reported in another investigation (75).
Concerning CDX2, in our article (10) we found there was an absence of expression in the squamous esophageal epithelium, as also found in other studies (76); however, it is possible that by employing a mRNA detection technique results may differ (77). The fact that this protein was expressed in almost all BE patients, with and without dysplasia, confirms the usefulness of this marker to identify BE, especially in doubtful cases. Particularly interesting was the difference in expression between LGD and HGD, as also reported in other authors' experience (78). In particular, there is an initial upregulation of CDX2 in intestinal metaplasia, followed by progressive decline in its expression from LGD to HGD to adenocarcinoma. However, on the basis of the two different types of dysplasia described in BE (18), related to separate gastric and intestinal pathways of carcinogenesis, in the literature this downward trend of CDX2 upregulation along the neoplastic progression was observed only in cases with adenomatous dysplasia but not in cases with foveolar dysplasia (79). This evidence suggests a role for CDX2 as a tumor suppressor in the metaplasia-dysplasia-carcinoma sequence in the intestinal but not in the gastric pathway. This different expression pattern of CDX2 could be due to different pathways leading to the malignancies arising in the two districts (80).
In normal esophageal tissue CDC2 was expressed in the parabasal layer (proliferative compartment) and occasionally in inflammatory elements of the lamina propria and the submucosa. A diffuse positivity for CDC2 expression was also found in ADC arisen in BE, with a significant progressive increase in relation to the degree of neoplastic differentiation, thus, this marker could find a useful application in distinguishing cases of BE without dysplasia from those associated with LGD or HGD (81). Moreover, CDC2 could play a role as a proliferation index and as potential target for potential therapeutic approaches aimed at blocking the CDC2/cyclin B complex, and thereby preventing the start of cellular mitosis. In conclusion regarding the analysis of COX-2, CDX2 and CDC2 in cancer progression, this study shows that particularly CDC2 (a marker with potential value for the identification of intraepithelial neoplasms), might have some role in the clinical-pathological assessment of patients with BE (10) (Fig. 4).
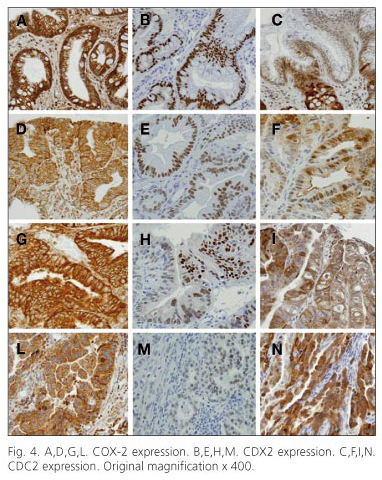
Recently it was demonstrated that enhancing the expression of CDX2 by stimulation with bile acids it was possible induce intestinal differentiation of esophageal columnar cells by interaction with the Notch signaling pathway (82). Furthermore, a correlation with the apical sodium-dependent bile acid transporter (ASBT) expression was found for CDX1, CDX2, and HNF-1α in BE biopsies, which suggests the human ASBT promoter is activated transcriptionally by CDX1 and CDX2 and provides a possible explanation for the reported observation that ASBT is aberrantly expressed in esophageal metaplasia that also expresses CDX transcription factors (83).
Conclusion
The ultimate goal of cancer surveillance and prevention programs is to identify those patients at high risk of developing a certain cancer, in order to treat the precursory condition, or to identify the invasive cancer at a very early stage. Another aim in cancer research and treatment is to combine the new findings in tumor markers studies and advanced treatment and therapy. Concerning esophageal ADC, the identification of intestinal metaplasia, i.e. BE and dysplasia, represents the major risk factor. Patients with BE have an annual incidence of esophageal carcinoma between 0.5 and 1%. Therefore, the histopathological classification of the grade of dysplasia in BE is the only currently accepted method for risk stratification of patients. However, progression from one lesion to another and lastly to cancer typically occurs with a long latency and shows considerable inter-individual heterogeneity. The risk factors are well known but the real impact of molecular markers is a knowledge which should be better understood. Thus, the identification of different dysplastic conditions is a poor predictor for how rapidly a patient will ultimately progress to cancer and the search for new markers useful for patients' treatment is desirable and strongly suggested.
Acknowledgments
We thank Ms. Sarah Owens for English revision.
References
1. Blot WJ, Devesa SS, Kneller RW, Fraumeni JF Jr. Rising incidence of adenocarcinoma of the esophagus and gastric cardia. JAMA 1991;265:1287-9. [ Links ]
2. Wang KK, Sampliner RE. Practice Parameters Committee of the American College of Gastroenterology. Updated guidelines 2008 for the diagnosis, surveillance and therapy of Barrett's esophagus. Am J Gastoenterol 2008;103:788-97. [ Links ]
3. Takubo K, Aida J, Naomoto Y, Sawabe M, Arai T, Shiraishi H, et al. Cardiac rather than intestinal-type background in endoscopic resection specimens of minute Barrett adenocarcinoma. Hum Pathol 2009;40:65-74. [ Links ]
4. Riddell RH, Odze RD. Definition of Barrett's esophagus: Time for a rethink - Is intestinal metaplasia dead? Am J Gastroenterol 2009;104:2588-94. [ Links ]
5. Hahn HP, Blount PL, Ayub K, Das KM, Souza R, Spechler S, et al. Intestinal differentiation in metaplastic, non-goblet columnar epithelium in the esophagus. Am J Surg Pathol 2009;33:1006-15. [ Links ]
6. Tosh D, Slack JM. How cells change their phenotype. Nat Rev Mol Cell Biol 2002;3:187-94. [ Links ]
7. Greenawalt DM, Duong C, Smyth GK, Ciavarella ML, Thompson NJ, Tiang T, et al. Gene expression profiling of esophageal cancer: Comparative analysis of Barrett's esophagus, adenocarcinoma, and squamous cell carcinoma. Int J Cancer 2007;120:1914-21. [ Links ]
8. Wild CP, Hardie LJ. Reflux, Barrett's oesophagus and adenocarcinoma: Burning questions. Nature Reviews Cancer 2003;3:676-84. [ Links ]
9. Jenkins GJ, Doak SH, Parry JM, D'Souza FR, Griffiths AP, Baxter JN. Genetic pathways involved in the progression of Barrett's metaplasia to adenocarcinoma. British J of Surgery 2002;89:824-37. [ Links ]
10. Villanacci V, Rossi E, Zambelli C, Galletti A, Cestari R, Missale G, et al. COX-2, CDX2, and CDC2 immunohistochemical assessment for dysplasia-carcinoma progression in Barrett's esophagus. Dig Liver Dis 2007;39:305-11. [ Links ]
11. Villanacci V, Bellone G, Battaglia E, Rossi E, Carbone A, Prati A, et al. Ski/SnoN expression in the sequence metaplasia-dysplasia-adenocarcinoma of Barrett's esophagus. Hum Pathol 2008;9:403-9. [ Links ]
12. Dunn JM, Mackenzie GD, Oukrif D, Mosse CA, Banks MR, Thorpe S, et al. Image cytometry accurately detects DNA ploidy abnormalities and predicts late relapse to high-grade dysplasia and adenocarcinoma in Barrett's oesophagus following photodynamic therapy. Br J Cancer 2010;102:1608-17. [ Links ]
13. Reid BJ. Barrett's esophagus and esophageal adenocarcinoma. Gastroenterol Lin North Am 1991;20:817-34. [ Links ]
14. Cestari R, Villanacci V, Rossi E, Della Casa D, Missale G, Conio M, et al. Fluorescence in situ hybridization to evaluate dysplasia in Barrett's esophagus: A pilot study. Cancer Lett 2007;251:278-87. [ Links ]
15. Rossi E, Villanacci V, Bassotti G, Casa DD, Missale G, Minelli L, et al. Her-2/neu in barrett esophagus: a comparative study between histology, immunohistochemistry, and fluorescence in situ hybridization. Diagn Mol Pathol 2006;15:125-30. [ Links ]
16. Sabo E, Meitner PA, Tavares R, Corless CL, Lauwers GY, Moss SF, et al. Expression analysis of Barrett's esophagus-associated high-grade dysplasia in laser capture microdissected archival tissue. Clin Cancer Res 2008;14:6440-8. [ Links ]
17. Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007;449:1003-7. [ Links ]
18. Brown IS, Whiteman DC, Lauwers GY. Foveolar type dysplasia in Barrett esophagus. Mod Pathol 2010;23:834-43. [ Links ]
19. Alikhan M, Rex D, Khan A, Rahmani E, Cummings O, Ulbright TM. Variable pathologic interpretation of columnar lined esophagus by general pathologists in community practice. Gastrointest Endosc 1999;50:23-6. [ Links ]
20. Galipeau PC, Cowan DS, Sanchez CA, Barrett MT, Emond MJ, Levine DS, et al. 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett's esophagus. Proc Natl Acad Sci U S A 1996;93:7081-4. [ Links ]
21. Rygiel AM, Milano F, Ten Kate FJ, de Groot JG, Peppelenbosch MP, Bergman JJ, et al. Assessment of chromosomal gains as compared to DNA content changes is more useful to detect dysplasia in Barrett's esophagus brush cytology specimens. Genes Chromosomes Cancer 2008;47:396-404. [ Links ]
22. Kerkhof M, Steyerberg EW, Kusters JG, van Dekken H, van Vuuren AJ, Kuipers EJ, et al. Aneuploidy and high expression of p53 and Ki67 is associated with neoplastic progression in Barrett esophagus. Cancer Biomark 2008;4:1-10. [ Links ]
23. Bailey T, Biddlestone L, Shepherd N, Barr H, Warner P, Jankowski J. Altered cadherin and catenin complexes in the Barrett's esophagus-dysplasia-adenocarcinoma sequence: Correlation with disease progression and differentiation. Am J Pathol 1998;152:135-44. [ Links ]
24. van Dekken H, Hop WC, Tilanus HW, Haringsma J, van der Valk H, Wink JC, et al. Immunohistochemical evaluation of a panel of tumor cell markers during malignant progression in Barrett esophagus. Am J Clin Pathol 2008;130:745-53. [ Links ]
25. Scheil-Bertram S, Lorenz D, Ell C, Sheremet E, Fisseler-Eckhoff A. Expression of alpha-methylacyl coenzyme A racemase in the dysplasia carcinoma sequence associated with Barrett's esophagus. Mod Pathol 2008;21:961-7. [ Links ]
26. Lagergren J, Bergstrom R, Lindgren A, Nyrén O. Symptomatic gastroesophageal reflux as a risk factor for esophageal ADC. N Engl J Med 1999;340:825-31. [ Links ]
27. Spechler SJ. Clinical practice. Barrett's esophagus. N Engl J Med 2002;346:836-42. [ Links ]
28. Kubo A, Corley DA. Body mass index and adenocarcinomas of the esophagus or gastric cardia: A systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev 2006;15:827-78. [ Links ]
29. El Serag HB, Kvapil P, Hacken-Bitar J, Kramer JR. Abdominal obesity and the risk of Barrett's esophagus. Am J Gastroenterology 2005;100:2151-6. [ Links ]
30. Rokkas T, Pistiolas D, Sechopoulos P, Robotis I, Margantinis G. Relationship between Helicobacter Pylori infection and esophageal neoplasia: A meta-analysis. Clin Gastroenterol Hepatol 2007;5:1413-7. [ Links ]
31. Arber N, Lightdale C, Rotterdam H, Han KH, Sgambato A, Yap E, et al. Increased expression of the cyclin D1 gene in Barrett's esophagus. Cancer Epidemiol Biomarkers Prev 1996;5:457-9. [ Links ]
32. Sarbia M, Bektas N, Müller W, Heep H, Borchard F, Gabbert HE. Expression of Cyclin E in dysplasia, carcinoma, and non-malignant lesions of Barrett esophagus. Cancer 1999;86:2597-601. [ Links ]
33. Geddert H, Heep HJ, Gabbert HE, Sarbia M. Expression of cyclin B1 in the metaplasia-dysplasia-carcinoma sequence of Barrett esophagus. Cancer 2002;94:212-8. [ Links ]
34. Lao-Sirieix, Lovat L, Fitzgerald RC. Cyclin A immunocytology as a risk stratification tool for Barrett's esophagus surveillance. Clin Cancer Res 2007;13:659-65. [ Links ]
35. Jankowski J, Coghill G, Hopwood D, Wormsley KG. Oncogenes and oncosuppressor gene in adenocarcinoma of the oesophagus. Gut 1992;33:1033-8. [ Links ]
36. Brito MJ, Filipe MI, Linehan J, Jankowski J. Association of transforming growth factor alpha (TGFA) and its precursors with malignant change in Barrett's epithelium: Biological and clinica variables. Int J Cancer 1995;60:27-32. [ Links ]
37. Lord RV, O'Grady R, Sheehan C, Field AF, Ward RL. K-ras codon 12 mutations in Barrett's oesophagus and adenocarcinomas of the oesophagus and oesophagogastric junction. J Gastroenterol Hepatol 2000;15:730-6. [ Links ]
38. Wong DJ, Barrett MT, Stoger R, Emond MJ, Reid BJ. p16INK4a promoter is hypermethylated at a high frequency in esophageal adenocarcinomas. Cancer Res 1997;57:2619-22. [ Links ]
39. Wong DJ, Paulson TG, Prevo LJ, Galipeau PC, Longton G, Blount PL, et al. p16(INK4a) lesions are common, early abnormalities that undergo clonal expansion in Barrett's metaplatic espithelium. Cancer Res 2001;61:8284-9. [ Links ]
40. Hetts SW. To die or not to die: An overview of apoptosis and its role in disease. JAMA 1998;279:300-07. [ Links ]
41. Popnikolov NK, Gatalica Z, Adegboyega PA, Norris BA, Pasricha PJ. Down regulation of TNF-related apoptosis-inducing ligand (TRAIL)/Apo2L in Barrett's esophagus with dysplasia and adenocarcinoma. Appl. Immunohistochem Mol Morphol 2006;14:161-5. [ Links ]
42. Souza RF, Shewmake K, Beer DG, Cryer B, Spechler SJ. Selective inhibition of cyclooxygenase-2 suppresses growth and induces apoptosis in human esophageal adenocarcinoma cells. Cancer Res 2000;60:5767-72. [ Links ]
43. Shirvani VN, Ouatu-Lascar R, Kaur BS, Omary MB, Triadafilopoulos G. Cyclooxygenase 2 expression in BE and adenocarcinoma: Ex vivo induction by bile salts and acid exposure. Gastroenterology 2000;118:487-96. [ Links ]
44. Morales CP, Lee EL, Shay JW. In situ hybridization for the detection of telomerase RNA in the progression from Barrett's esophagus to esophageal adenocarcinoma. Cancer 1998; 83:652-9. [ Links ]
45. López-Novoa JM, Bernabeu C. The physiological role of endoglin in the cardiovascular system. Am J Physiol Heart Circ Physiol 2010;299:H959-74. [ Links ]
46. Bernabeu C, Lopez-Novoa JM. The emerging role of TGF-beta superfamily coreceptors in cancer. Quintanilla M Biochim Biophys Acta 2009;1792:954-73. [ Links ]
47. Saad RS, El-Gohary Y, Memari E, Liu YL, Silverman JF. Endoglin (CD105) and vascular endothelial growth factor as prognostic markers in esophageal adenocarcinoma. Hum Pathol 2005;36:955-61. [ Links ]
48. Bailey T, Biddlestone L, Shepherd N, Barr H, Warner P, Jankowski J. Altered cadherin and catenin complexes in the Barrett's esophagus-dysplasia-adenocarcinoma sequence: Correlation with disease progression and differentiation. Am J Pathol 1998;152:135-44. [ Links ]
49. El Serag HB, Aguirre TV, Davis S, Kuebeler M, Bhattacharyya A, Sampliner RE. Proton Pump Inhibitors Are Associated with Reduced Incidence of Dysplasia in Barrett's Esophagus. Am J Gastroenterol 2004;99:1877-83. [ Links ]
50. Pandolfino JE, Kahrilas PJ. American Gastroenterological Association. American Gastroenterological Association medical position statement: Clinical use of esophageal manometry. Gastroenterology 2005;128:207-8. [ Links ]
51. DeVault KR, Castell DO, American College of Gastroenterology. Updated Guidelines for the Diagnosis and Treatment of Gastroesophageal Reflux Disease. Am J Gastroenterol 2005;100:190-200. [ Links ]
52. Ross JS, Fletcher JA. The HER-2/neu oncogene: Prognostic factor, predictive factor and target for therapy. Semin Cancer Biol 1999;9:125-38. [ Links ]
53. Werner M, Mueller J, Walch A, Höfler H. The molecular pathology of Barrett's esophagus. Histol Histopathol 1999;14:553-9. [ Links ]
54. Bianco AR. Targeting c-erbB2 and other receptors of the c-erbB family: Rationale and clinical applications. J Chemother 2004;16:52-4. [ Links ]
55. Masood S, Bui MM. Prognostic and predictive value of HER2/neu oncogene in breast cancer. Microsc Res Tech 2002;59:102-8. [ Links ]
56. Fléjou JF, Paraf F, Muzeau F, Fékété F, Hénin D, Jothy S, et al. Expression of c-erbB-2 oncogene product in Barrett's adenocarcinoma: pathological and prognostic correlations. J Clin Pathol 1994;47:23-6. [ Links ]
57. Walch A, Specht K, Bink K, Zitzelsberger H, Braselmann H, Bauer M, et al. Her-2/neu gene amplification, elevated mRNA expression, and protein overexpression in the metaplasia-dysplasia-adenocarcinoma sequence of Barrett's esophagus. Lab Invest 2001;81:791-801. [ Links ]
58. Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr, Davidson NE, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med 2005;353:1673-84. [ Links ]
59. Hardwick RH, Shepherd NA, Moorghen M, Newcomb PV, Alderson D. c-erbB-2 overexpression in the dysplasia/carcinoma sequence of Barrett's oesophagus. J Clin Pathol 1995;48:129-32. [ Links ]
60. Rossi E, Grisanti S, Villanacci V, Della Casa D, Cengia P, Missale G, et al. HER-2 overexpression/amplification in Barrett's oesophagus predicts early transition from dysplasia to adenocarcinoma: A clinico-pathologic study. J Cell Mol Med 2009;13:3826-33. [ Links ]
61. Weston AP, Sharma P, Mathur S, Banerjee S, Jafri AK, Cherian R, et al. Risk stratification of Barrett's esophagus: Updated prospective multivariate analysis. Am J Gastroenterol 2004;99:1657-66. [ Links ]
62. Rossi E, Villanacci V, Danesino C, Donato F, Nascimbeni R, Bassotti G. Epidermal growth factor receptor overexpression/amplification in adenocarcinomas arising in the gastrointestinal tract. Rev Esp Enferm Dig 2011;103:632-9. [ Links ]
63. Järvinen TA, Liu ET. HER-2/neu and topoisomerase IIalpha - Simultaneous drug targets in cancer. Comb Chem High Throughput Screen 2003;6:455-70. [ Links ]
64. Rossi E, Villanacci V, Bassotti G, Donato F, Festa A, Cengia G, et al. TOPOIIalpha and HER-2/neu overexpression/amplification in Barrett's oesophagus, dysplasia and adenocarcinoma. Histopathology 2010;57:81-9. [ Links ]
65. Isaacs RJ, Davies SL, Wells NJ, Harris AL. Topoisomerases II alpha and beta as therapy targets in breast cancer. Anticancer Drugs 1995;6:195-211. [ Links ]
66. Murphy AJ, Hughes CA, Barrett C, Magee H, Loftus B, O'Leary JJ, et al. Low-level TOP2A amplification in prostate cancer is associated with HER2 duplication, androgen resistance, and decreased survival. Cancer Res 2007;15:2893-8. [ Links ]
67. Lin L, Bass AJ, Lockwood WW, Wang Z, Silvers AL, Thomas DG, et al. Activation of GATA binding protein 6 (GATA6) sustains oncogenic lineage-survival in esophageal adenocarcinoma. Proc Natl Acad Sci U S A 2012;109:4251-6. [ Links ]
68. Buskens CJ, Van Rees BP, Sivula A, Reitsma JB, Haglund C, Bosma PJ, et al. Prognostic significance of elevated cyclooxygenase 2 expression in patients with adenocarcinoma of the esophagus. Gastroenterology 2002;122:1800-7. [ Links ]
69. Tuynman JB, Buskens CJ, Kemper K, ten Kate FJ, Offerhaus GJ, Richel DJ, et al. Neoadjuvant selective COX- 2 inhibition down-regulates important oncogenic pathways in patients with esophageal adenocarcinoma. Ann Surg 2005;242:840-50. [ Links ]
70. Scholl C, Bansal D, Döhner K, Eiwen K, Huntly BJ, Lee BH, et al. The homeobox gene CDX2 is aberrantly expressed in most cases of acute myeloid leukemia and promotes leukemogenesis. J Clin Invest 2007;117:1037-48. [ Links ]
71. Silberg DG, Swain GP, Suh ER, Traber PG. CDX1 and CDX2 expression during intestinal development. Gastroenterology 2000;119:961-71. [ Links ]
72. Nozoe T, Takahashi I, Baba H, Maehara Y. Relationship between intracellular localization of p34cdc2 protein and differentiation of esophageal squamous cell carcinoma. J Cancer Res Clin Oncol 2005;131:179-83. [ Links ]
73. Kandil HM, Tanner G, Smalley W, Halter S, Radhika A, Dubois RN. Cyclooxygenase-2 expression in Barrett's esophagus. Dig Dis Sci 2001;46:785-9. [ Links ]
74. Abdalla SI, Lao-Sirieix P, Novelli MR, Lovat LB, Sanderson IR, Fitzgerald RC. Gastrin-induced Ciclooxygenase-2 expression in Barrett's carcinogenesis. Clin Cancer Res 2004;10:4784-92. [ Links ]
75. Morris CD, Armstrong GR, Bigley G, Green H, Attwood SE. Cyclooxygenase-2 expression in the Barrett's metaplasia-dysplasia-adenocarcinoma sequence. Am J Gastroenterol 2001;96:990-6. [ Links ]
76. Eda A, Osawa H, Satoh K, Yanaka I, Kihira K, Ishino Y, et al. Aberrant expression of CDX2 in Barrett's epithelium and inflammatory esophageal mucosa. J Gastroenterol 2003;38:14-22. [ Links ]
77. Moons LM, Bax DA, Kuipers EJ, Van Dekken H, Haringsma J, Van Vliet AH, et al. The homeodomain protein CDX2 is an early marker of Barrett's oesophagus. J Clin Pathol 2004;57:1063-8. [ Links ]
78. Kaimaktchiev V, Terracciano L, Tornillo L, Spitchin H, Stoios D, Bundi M, et al. The homeobox intestinal differentiation factor CDX2 is selectively expressed in gastrointestinal adenocarcinomas. Mod Pathol 2004;17:1392-9. [ Links ]
79. Khor TS, Alfaro EE, Ooi EM, Li Y, Srivastava A, Fujita H, et al. Divergent expression of MUC5AC, MUC6, MUC2, CD10, and CDX-2 in dysplasia and intramucosal adenocarcinomas with intestinal and foveolar morphology: Is this evidence of distinct gastric and intestinal pathways to carcinogenesis in Barrett Esophagus? Am J Surg Pathol 2012;36:331-42. [ Links ]
80. Cengia G, Missale G, Minelli L, Villanacci V, Rossi E, Cestari R. Screening for and surveillance of Barrett's esophagus is clinically indicated. Dig Dis 2007;25:197-202. [ Links ]
81. Hansel DE, Dhara S, Huang RC, Ashfaq R, Deasel M, Shimada Y, et al. CDC2/CDK1 expression in esophageal adenocarcinoma and precursor lesions serves as a diagnostic and cancer progression marker and potential novel drug target. Am J Surg Pathol 2005;29:390-9. [ Links ]
82. Tamagawa Y, Ishimura N, Uno G, Yuki T, Kazumori H, Ishihara S, et al. Notch signaling pathway and Cdx2 expression in the development of Barrett's esophagus. Lab Invest 2012; 92:896-909. [ Links ]
83. Ma L, Jüttner M, Kullak-Ublick GA, Eloranta JJ. Regulation of the gene encoding the intestinal bile acid transporter ASBT by the caudal-type homeobox proteins CDX1 and CDX2. Am J Physiol Gastrointest Liver Physiol 2012;302:G123-33. [ Links ]
![]() Correspondence:
Correspondence:
Elisa Rossi.
Centro de Investigaciones Biológicas.
c/ Ramiro de Maeztu, 9.
28040 Madrid, Spain
e-mail: e.rossi@cib.csic.es
Received: 29-09-2012
Accepted: 17-12-2012

