










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versão impressa ISSN 1130-0108
Rev. esp. enferm. dig. vol.109 no.2 Madrid Fev. 2017
https://dx.doi.org/10.17235/reed.2016.4224/2016
Manejo del gastrinoma pancreático asociado a la enfermedad de Von Hippel-Lindau. A propósito de un caso
Management of pancreatic gastrinoma associated with Von Hippel-Lindau disease: a case report
Ángela Sala-Hernández, Eva María Montalvá Orón, Eugenia Pareja Ibars, Neus Ballester Pla y Rafael López-Andújar
Departamento de Cirugía General. Hospital Universitario y Politécnico La Fe. Valencia
Dirección para correspondencia
RESUMEN
Introducción: los tumores neuroendocrinos de páncreas (TNEP) son un grupo heterogéneo y constituyen el 1,3% de todos los tumores pancreáticos. Aproximadamente el 10% aparecen en el contexto de síndromes familiares como el Von Hippel-Lindau (VHL).
Caso clínico: presentamos el caso de una paciente mujer de 37 años diagnosticada de VHL e intervenida en varias ocasiones por hemangioblastomas cerebrales y carcinomas renales. Durante su seguimiento se diagnostica de 2 gastrinomas funcionantes menores de 2 cm que se enuclearon. Posteriormente desarrolló nuevo TNEP y se le realizó una duodenopancreatectomía total sin preservación pilórica.
Discusión: el manejo de los TNEP en el VHL es difícil debido a la asociación de múltiples tumores en diferentes órganos y a la morbi-mortalidad asociada a la cirugía del páncreas. Su tratamiento hay que individualizarlo en cada paciente, basándonos en su capacidad de producción de hormonas y, por tanto de dar sintomatología, en su tamaño y localización y, además debe ser contextualizado con el resto de tumores que suelen presentar estos pacientes.
Palabras clave: Von Hippel-Lindau. Tumor neuroendocrino. Páncreas. Gastrinoma.
ABSTRACT
Background: Pancreatic neuroendocrine tumors (PNET) are a heterogeneous group and constitute 1.3% of all pancreatic tumors. Approximately 10% of these occur in the context of hereditary syndromes, such as VHL disease.
Case report: We report a case of a female patient of 37 years diagnosed VHL and intervened on several occasions by cerebral hemangioblastoma and renal carcinomas. During its follow-up she was diagnosed 2 gastrinomas functioning under 2 cm were enucleated. Later developed new PNET and underwent a total duodenopancreatectomy without pyloric preservation.
Discussion: The management of PNET in VHL is difficult due to the association of multiple tumors in different organs and the morbidity and mortality associated with the surgery of the pancreas. Management must be individualized for each patient, based on the ability to produce hormones and present symptoms, the size and location, and in the context of other tumors that usually present in these patients.
Key words: Von Hippel-Lindau. Neuroendocrine tumour. Pancreas. Gastrinoma.
Introducción
La enfermedad de Von Hippel-Lindau (VHL) es una enfermedad rara, de carácter hereditario autosómico dominante, caracterizada por el desarrollo de tumores y quistes en diferentes órganos.
Los órganos en los que con más frecuencia asientan los tumores son: riñón (carcinoma de células claras) (25-60%), glándula adrenal (feocromocitoma) (10-20%), sistema nervioso central (hemangioblastoma) (44-72%), ojo (angioma retiniano) (25-60%), oído interno (neoplasia del saco endolinfático) (10-25%), epidídimo (cistoadenoma) (25-60%) y páncreas (tumores neuroendocrinos y quistes) (35-77%) (1,2).
Los quistes serosos representan la lesión más frecuente en el páncreas (70%) (3), mientras que los tumores neuroendocrinos lo afectan entre un 10-17% (4), siendo generalmente únicos y no funcionantes.
A diferencia de los tumores neuroendocrinos de páncreas (TNEP) esporádicos no funcionantes y con alto potencial de malignidad (60-100%), los TNEP relacionados con el síndrome VHL tienen una tasa mucho menor de metástasis. Dado que no producen secreción hormonal, no dan síntomas y son, por tanto, diagnosticados en estadios avanzados. Habitualmente los pacientes que padecen el síndrome de VHL son sometidos desde la infancia a pruebas de screening, por lo que se realiza un diagnóstico temprano de los TNEP (5).
El objetivo de esta nota clínica es presentar un caso clínico de TNEP funcionante y maligno en una paciente con diagnóstico de Von Hippel-Lindau y páncreas multiquístico.
Caso clínico
Paciente mujer de 37 años diagnosticada de VHL, portadora de una derivación ventrículo peritoneal. Intervenida en dos ocasiones por carcinomas renales (resección parcial renal derecha y una radiofrecuencia de un nódulo en el riñón izquierdo), y en cuatro ocasiones se le realizó exéresis de hemangioblastomas cerebrales.
Durante el seguimiento se detectaron, mediante tomografía por emisión de positrones-tomografía computarizada (PET-TC), dos lesiones en cabeza del páncreas con valor máximo estandarizado de captación (SUV máx.) 14,94 g/ml y múltiples quistes pancreáticos (Fig. 1). A continuación, se realizó un estudio funcional pancreático en el que se evidenció una gastrina sérica elevada (2.319 pg/ml). La punción-aspiración con aguja fina (PAAF) transgástrica de una de las lesiones sólidas mostró una citología compatible con tumor neuroendocrino de páncreas.
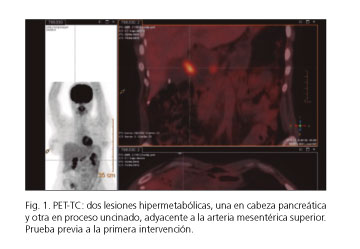
Con el diagnóstico de 2 gastrinomas funcionantes menores de 2 cm (T1N0M0 estadio IA, con grado de diferenciación G2, menos de 2 mitosis por campo y Ki-67 2-5%) se indicó cirugía conservadora en el comité multidisciplinar (6).
Durante la intervención, tras una exposición amplia del páncreas, se localizaron con ecografía las dos lesiones sólidas en la cabeza y proceso uncinado pancreático, que fueron enucleadas con disector ultrasónico (CUSA) y bisturí armónico, sin incidencias. El postoperatorio transcurrió sin complicaciones.
El estudio anatomopatológico puso en evidencia dos tumores neuroendocrinos moderadamente diferenciados (G2) (cromogranina, sinaptofisina y CD 56 +), con un índice de proliferación celular (Ki-67) del 10% y figuras mitóticas atípicas (1 mitosis en 10 campos de gran aumento). La gastrina postoperatoria mostró un descenso importante pero sin llegar a rango de normalidad (1.383 pg/ml).
A los 7 meses de seguimiento se objetivó nueva elevación de este biomarcador y una determinación elevada de cromogranina A (181 ng/ml).
Aunque el octreoscan no advertía acúmulo anormal de actividad, la resonancia magnética (RM) y el PET-TC objetivaron, además de crecimiento del tumor en riñón izquierdo tratado previamente con radiofrecuencia, nueva lesión en cabeza de páncreas en contacto con la vena mesentérica superior (Fig. 2).
Ante los hallazgos, y en el contexto de un páncreas multiquístico, se decidió en comité multidisciplinar una duodenopancreatectomía total sin preservación pilórica asociada a esplenectomía y resección del tumor del polo superior del riñón izquierdo. Para la reconstrucción del tránsito intestinal se confeccionó una gastroyeyunostomía término-lateral transmesocólica y una hepático-yeyunostomía término-lateral en Y de Roux. Al no identificar plano de separación entre vena porta y páncreas se realizó una resección parcial lateral de porta que se reconstruyó con sutura continua. La biopsia extemporánea descartó infiltración tumoral del vaso (Fig. 3).
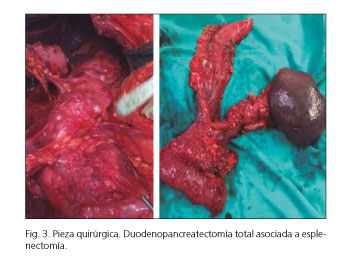
El informe histológico describió tumor neuroendocrino pancreático de 9 mm en el proceso uncinado de moderada diferenciación sin infiltración ganglionar, múltiples formaciones quísticas en todo el páncreas (cistoadenomas serosos multiquísticos y multifocales, con áreas de adenomas serosos-sólidos), ausencia de infiltración tumoral ganglionar (0/8) y carcinoma renal de células claras de 2,8 cm.
En el postoperatorio, la paciente presentó una fístula urinaria calicial que requirió drenaje por radiología intervencionista, resuelta tras 2 meses de seguimiento (complicación grado III de la clasificación de Clavien-Dindo).
Discusión
Los tumores neuroendocrinos de páncreas son un grupo heterogéneo y constituyen el 1,3% de todos los tumores pancreáticos.
Aproximadamente el 10% aparece en el contexto de síndromes familiares como el VHL. En la enfermedad de VHL los TNEP tienen mejor pronóstico que los TNEP esporádicos (enfermedad metastásica 10-20% frente al 60-90%, respectivamente).
Estas neoplasias pueden presentar un comportamiento maligno (5,7). Clínicamente pueden ser o no funcionantes. Mientras que las primeras dan manifestaciones clínicas relacionadas con las hormonas secretadas, las segundas se suelen manifestar por la compresión que ejercen sobre estructuras anatómicas vecinas o son detectadas como "incidentalomas" en el transcurso de exploraciones realizadas por otras patologías.
Para la evaluación y diagnóstico de los TNEP se deben realizar un test de determinación hormonal, pruebas de imagen y estudio histopatológico.
La determinación de los niveles plasmáticos de marcadores bioquímicos se ha convertido en una herramienta esencial no sólo en el diagnóstico sino también en el seguimiento de los pacientes con tumores neuroendocrinos funcionantes.
En este caso se realizó un estudio funcional que detectó elevación de gastrina, cuyo valor descendió tras la primera intervención, y volvió a incrementarse en controles posteriores, lo que sirvió para detectar una nueva recidiva.
Los estudios de imagen convencionales para la localización de los TNEP incluyen la ecografía endoscópica, la TC y la RM. Hoy en día, se dispone de ligandos marcados radiactivamente, que han supuesto un gran avance en el diagnóstico, localización y seguimiento de estos tumores. La gammagrafía con octreótido y el PET-TC (5) son exploraciones complementarias disponibles que ayudan a la localización de las lesiones. En esta paciente se determinó su localización en el PET-TC, mientras que el resultado del octreoscan resultó ser un falso negativo.
El manejo de los TNEP en el VHL es difícil debido a la asociación de múltiples tumores en diferentes órganos y a la morbimortalidad asociada a la cirugía del páncreas.
En los tumores asintomáticos, hay autores que se basan en el tamaño y la localización de la lesión, recomendando realizar seguimiento con TC con contraste o RM cada 12 meses cuando las lesiones son menores de 1 cm. En las lesiones con tamaño entre 1 y 3 cm, el manejo depende de su localización; así, está indicada la resección quirúrgica en aquellos tumores mayores de 2 cm localizados en la cabeza pancreática y a partir de 3 cm cuando se encuentran en el cuerpo (4).
En los tumores funcionantes, el tratamiento recomendado es la resección quirúrgica, independientemente de su tamaño y localización (2,9).
Blansfield y cols. (4) proponían un manejo dependiendo de la probabilidad de enfermedad metastásica basándose en tres factores: la presencia de mutación en el exón 3, el tiempo de duplicación del tamaño en menos de 500 días y el tamaño mayor de 3 cm. Aquellos pacientes sin ningún factor tenían poca probabilidad de enfermedad metastásica y en ellos se aconsejaba seguimiento radiológico cada 2 o 3 años; si el paciente presentaba uno de estos criterios, la vigilancia debía realizarse cada 6 o 12 meses, y si cumplían dos o tres, implicaba un alto potencial metastásico y recomendaban la resección quirúrgica (10).
Las opciones de tratamiento quirúrgico oscilan desde la enucleación, para aquellos tumores pequeños o de bajo grado alejados del conducto pancreático, hasta resecciones parciales o totales (9).
El tratamiento recomendado en caso de recurrencia tumoral, como ocurrió en esta paciente, es la cirugía cuando la enfermedad es susceptible de ser resecada completamente.
Conclusión
Los tumores neuroendocrinos funcionantes de páncreas asociados con el síndrome VHL son lesiones poco frecuentes. Su tratamiento hay que individualizarlo en cada paciente, basándonos en su capacidad de producción de hormonas y, por tanto de dar sintomatología, en su tamaño y localización y, además debe ser contextualizado con el resto de tumores que suelen presentar estos pacientes.
![]() Dirección para correspondencia:
Dirección para correspondencia:
Ángela Sala Hernández.
Departamento de Cirugía General.
Hospital Universitario Politécnico La Fe.
Avda. de Fernando Abril Martorell, 106.
46026 Valencia, Spain
e-mail: asalahdez@gmail.com
Recibido: 29-01-2016
Aceptado: 25-02-2016
Bibliografía
1. Eras M1, Yenigun M, Acar C, et al. Pancreatic involvement in Von Hippel-Lindau disease. Indian J Cancer 2004; 41:159-61. [ Links ]
2. Lonser RR, Glenn GM, Walther M, et al. Von Hippel-Lindau disease. Lancet 2003;361:2059-67. DOI: 10.1016/S0140-6736(03)13643-4. [ Links ]
3. Hammel PR, Vilgrain V, Terris B, et al.Pancreatic involvement in von Hippel-Lindau disease. The Groupe Francophone d'Etude de la Maladie de von Hippel-Lindau. Gastroenterology 2000;119:1087-95. DOI: 10.1053/gast.2000.18143. [ Links ]
4. Blansfield JA, Choyke L, Morita SY, et al. Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine tumors. Surgery 2007;142:814-8. DOI: 10.1016/j.surg.2007.09.012. [ Links ]
5. Ro C1, Chai W, Yu VE, et al. Pancreatic neuroendocrine tumors: biology, diagnosis, and treatment. Chin J Cancer 2013;32:312-24. DOI: 10.5732/cjc.012.10295. [ Links ]
6. Klöppel G, Rindi G, Perren A, et al. The ENETS and AJCC/UICC TNM classifications of the neuroendocrine tumors of the gastrointestinal tract and the pancreas: a statement. Virchows Arch 2010;456(6):595-7. [ Links ]
7. Libutti SK, Choyke PL, Bartlett DL, et al. Pancreatic neuroendocrine tumors associated with von Hippel Lindau disease: diagnostic and management recommendations. Surgery 1998;124:1153-9. DOI: 10.1067/msy.1998.91823. [ Links ]
8. Tan EH, Tan CH. Imaging of gastroenteropancreatic neuroendocrine tumors. World J Clin Oncol 2011;2:28-43. DOI: 10.5306/wjco.v2.i1.28. [ Links ]
9. Charlesworth M, Verbeke CS, Falk GA, et al. Pancreatic lesions in von Hippel-Lindau disease? A systematic review and meta-synthesis of the literature.J Gastrointest Surg 2012;16:1422-8. DOI: 10.1007/s11605-012-1847-0. [ Links ]
10. Kulke MH, Shah MH, Benson AB 3rd. Neuroendocrine tumors, version 1.2015. J Natl Compr Canc Netw 2015;13:78-108. [ Links ]
