










 Serviços customizados
Serviços customizados
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkPRESENTACIÓN
Varón de 54 años en seguimiento por el Servicio de Medicina Interna por su antecedente de feocromocitoma, operado en otro Centro hace 30 años con extirpación de ambas glándulas suprarrenales y seguimiento de su tensión arterial. Actualmente en tratamiento con labetalol e hidroaltesona. En la historia actual el paciente presenta frecuentes calambres en miembros superiores e inferiores, disestesias en pie derecho y dolor mecánico en dedos de la mano izquierda. En la exploración destaca TA 140 / 90 mmHg y hepatomegalia indolora y en la analítica GOT 57 U/L (Normal:0-37U/L), GPT 102 U/L (Normal:0-41U/L) y GGT 78 U/L (Normal:11-50U/L) y metanefrinas en orina elevadas: 1,7 mg/24h (Normal < 0,4), resto de analítica, incluida sodio, potasio, fósforo magnesio y CPK normal.
En el servicio de neurofisiología se le realizó un estudio de potenciales evocados somatosensoriales sin alteraciones.
Se realiza una TC abdominopélvica donde se identifican pequeñas imágenes nodulares hipervasculares en páncreas (figura 1) e intramedulares (figura 2) y varios pequeños nódulos renales, algunos quísticos y otros sólidos hipervasculares (figura 3), sugieren tumores neuroendocrinos pancreáticos, hemangioblastomas medulares y tumores de células renales.
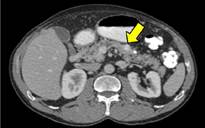
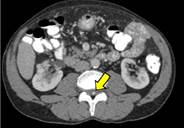
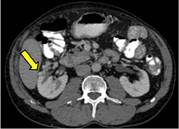
Se completa el estudio con RM de columna y cerebral, donde se comprueba la existencia de múltiples imágenes nodulares intraparenquimatosas en cerebelo y médula hipervasculares (figura 4 y 5), compatibles con hemangioblastomas.
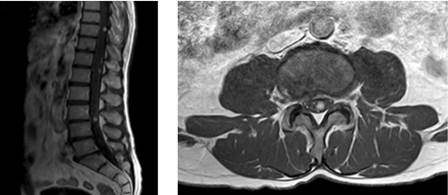
Se trata de una enfermedad multisistémica con afectación pancreática, renal y del sistema nervioso central en un paciente con antecedente de feocromocitoma intervenido mediante suprarrenalectomía bilateral, que mantiene tensión arterial elevada y niveles de metanefrinas en orina elevados.
DIAGNÓSTICO: SÍNDROME DE VON HIPPEL- LINDAU
Existen múltiples enfermedades sistémicas que presentan manifestaciones abdominales con presencia de tumores y seudotumores en varios órganos. Entre ellos destacamos las siguientes enfermedades congénitas:
Neurofibromatosis tipo 1 o enfermedad de von Recklinghausen1: Es un trastorno congénito neurocutáneo caracterizado por la presencia de neurofibromas cutáneos y plexiformes en el resto del organismo. Tienen mayor predisposición a desarrollar tumores neuroendocrinos, feocromocitomas y paragangliomas, tumores del estroma y adenocarcinomas gastrointestinales y neoplasias en el SNC.
Esclerosis tuberosa o enfermedad de Bourneville2: Es un síndrome neurocutáneo congénito que se caracteriza por la presencia de tumores en múltiples órganos, en el SNC, los hamartomas corticales son las lesiones más características, también se encuentran hamartomas y astrocitomas subependimarios. Fuera del SNC son frecuentes los angiofibromas y fibromas cutáneos, quistes y angiomiolipomas renales, carcinoma de células renales y adenomas esplénicos y pancreáticos.
Síndrome de Von Hippel- Lindau3: Es un trastorno neurocutáneo congénito caracterizado por la presencia de hemangioblastomas retinianos y en SNC y mayor predisposición que la población general a desarrollar feocromocitomas, quistes y tumores renales múltiples, y quistes, citoadenomas y tumores neuroendocrinos pancreáticos, aunque con una frecuencia menor, puede asociar tumores del saco endolinfático y cistoadenomas del epidídimo o del ligamento ancho.
Síndrome de neoplasias endocrinas múltiples4: Es un síndrome congénito que se caracteriza por la aparición de dos o más tumores neuroendocrinos. El síndrome MEN tipo IIA se caracteriza por la presencia de carcinoma medular de tiroides, feocromocitoma e hiperplasia paratiroidea y el IIB por presencia de carcinoma medular de tiroides, feocromocitoma y neuromas.
Sindrome del paraganglioma hereditario5: Es una enfermedad hereditaria caracterizada por la presencia de paragangliomas, tumores que crecen en los tejidos neuroendocrinos distribuidos simétricamente a lo largo del eje paravertebral desde el cráneo hasta la pelvis y feocromocitomas, paragangliomas que se limitan a la médula suprarrenal.
Feocromocitoma maligno6: Por último no se puede descartar un feocromocitoma maligno con depósitos secundarios, en un paciente en el que se palpa hepatomegalia y presenta hipertransaminasemia. Aunque la supervivencia del feocromocitoma maligno a los cinco años es del 40%, se pueden manifestar los depósitos secundarios varios años después del diagnóstico, principalmente en huesos, hígado y pulmón.
Se realizó al paciente un estudio genético donde se detectó en heterocigosis la variante compatible con Síndrome de Von Hippel-Lindau.
El Síndrome de Von Hippel-Lindau (VHL)7) es un trastorno multisistémico poco frecuente, que se transmite de forma autosómica dominante con alta penetrancia y expresión variable. Se manifiesta en edades tempranas de la vida y provoca una aparición desordenada de tumores malignos y benignos. El gen está localizado en el brazo corto del cromosoma 3, se produce una inactivación de un gen supresor de tumores.
Las manifestaciones clínicas más comunes son los hemangioblastomas del SNC y los angiomas retinianos, y el carcinoma renal y el feocromociroma son las principales causas potenciales de morbimortalidad.
Según el National Cancer Institute se clasifica en: Tipo I: VHL sin feocromocitoma. Es el más común. Hemangiobalstomas retinianos y del SNC, quistes y carcinomas renales y quistes pancreáticos. Tipo II: VHL con feocromocitoma. IIA: Hemangiobalstomas retinianos y del SNC, tumores pancreáticos y feocromocitoma. IIB: Hemangiobalstomas retinianos y del SNC, carcinoma de células renales, tumores pancreáticos y feocromocitoma.
El tratamiento de esta enfermedad es multidisciplinar y dependerá del tipo de tumor asociado. En muchas ocasiones se decide observación si las lesiones permanecen estables y asintomáticas. Las complicaciones varían en función del tumor subyacente, los tumores renales son la principal causa de muerte en estos pacientes, que por lo general tienen una esperanza de vida de 49 años.
Actualmente nuestro paciente se encuentra en seguimiento por parte del Servicio de Medicina Interna en colaboración con otros Servicios (Urología, Neurocirugía, Digestivo), realizándose periódicamente pruebas de imagen. Desde el diagnóstico hace 5 años los tumores pancreáticos y del SNC permanecen estables y los renales han crecido ligeramente, pero debido a su pequeño tamaño, menores de 1 cm, se están controlando a la espera de que si aumentan se traten quirúrgicamente o mediante ablación con radiofrecuencia.
