










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkNefrología (Madrid)
versão On-line ISSN 1989-2284versão impressa ISSN 0211-6995
Nefrología (Madr.) vol.32 no.6 Cantabria 2012
https://dx.doi.org/10.3265/Nefrologia.pre2012.Jul.11658
Poliquistosis renal autosómica dominante y agenesia renal contralateral
Autosomal dominant polycystic kidney disease with contralateral renal agenesis
Dirección para correspondencia
Sr. Director:
La poliquistosis renal autosómica dominante (PQRAD) es la causa hereditaria más frecuente de insuficiencia renal crónica (IRC) terminal, con una incidencia de 1 en 500-1000. La enfermedad está causada por mutaciones en los genes PKD1 (16p13.3, 85 %) o PKD2 (4q22.1, 15 %). En la PQRAD, el crecimiento de los quistes origina aumento progresivo del volumen renal y destrucción del parénquima, conduciendo a la IRC terminal a los 50-60 años (en la PKD1). Aunque la PQRAD es bilateral, la afectación renal puede ser asíncrona y asimétrica2, y en la PKD2 la IRC terminal puede retrasarse hasta 20 años. Por otra parte, las anomalías congénitas del riñón y del tracto urinario (CAKUT) son la causa más frecuente de IRC en el niño, contándose en la mitad de todos los casos3-6. Aunque muchas CACUK son causadas por defectos genéticos únicos, se han identificado mutaciones en solo unos pocos genes. Estas mutaciones únicas pueden causar un amplio espectro fenotípico de CAKUT, que van desde el reflujo vesicoureteral a la agenesia renal5-7. La agenesia renal unilateral es una anomalía congénita, que se presenta en 1 de 3000 nacidos3 y para la que existen varios genes candidatos. La coexistencia de PQRAD y agenesia renal unilateral puede ocurrir en 1 de 1.500.000-3.000.000 de individuos. Por lo tanto, la coincidencia de ambas anomalías es rara, con solo 7 casos comunicados8-12.
En una cohorte de 205 pacientes con PQRAD hemos identificado un nuevo caso de PQRAD con agenesia renal unilateral, que es el octavo caso comunicado en el mundo y el segundo en España. Se trata de un varón de 57 años que a los 41 fue diagnosticado de PQRAD derecha y agenesia renal izquierda por ecografía. Entonces presentaba creatinina sérica (Crs) de 1,2 mg/dl. La PQRAD derecha y la agenesia renal izquierda se confirmaron mediante renograma isotópico y angiorresonancia magnética (RM). La RM mostró quistes hepáticos, un gran riñón poliquístico derecho y ausencia de tejido renal en el lado izquierdo (figura 1). Dieciséis años después, la Crs fue 5,9 mg/dl y el aclaramiento de Cr 12 ml/min/1,73 m2. En los últimos 6 años la pérdida media anual del filtrado glomerular fue de 7,6 ml/min/1,73 m2, precisando diálisis peritoneal a partir de los 57 años. En los antecedentes familiares la madre, dos hermanos y uno de sus dos hijos tenían PQRAD (figura 2). El estudio familiar descartó la presencia de agenesia renal unilateral en otros miembros de la familia.
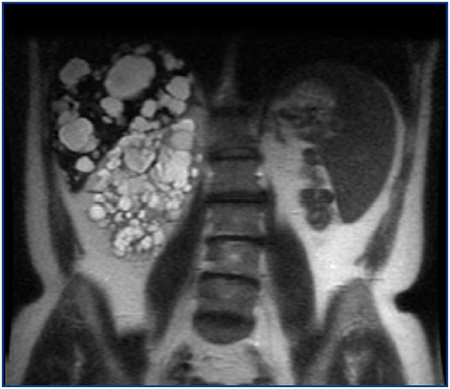
Figura 1. Imagen coronal de resonancia magnética mostrando
un gran riñón poliquístico derecho y ausencia de riñón del lado izquierdo.
El hígado muestra también múltiples quistes
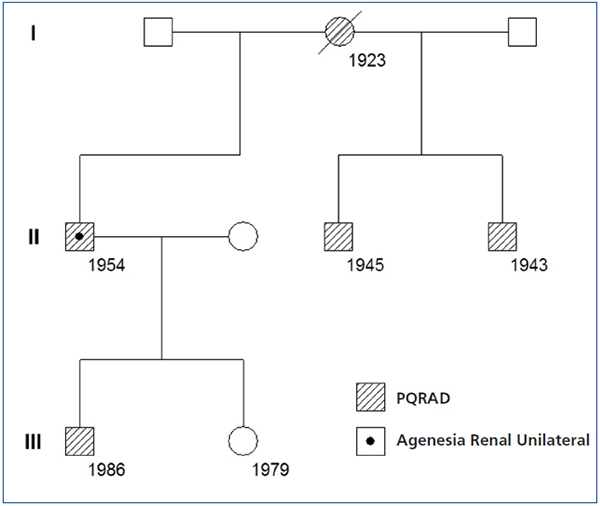
Figura 2. Árbol genealógico de la familia con poliquistosis renal autosómica dominante
y agenesia renal unilateral. Los números indican la fecha de nacimiento
PQRAD: poliquistosis renal autosómica dominante.
La agenesia renal se produce por la falta de interacción entre la yema ureteral y el mesénquima metanéfrico, originando la ausencia de uréter y riñón. La inducción de la yema ureteral desde el conducto néfrico es mediada por el factor neurotrófico derivado de la glia (GDNF), secretado por el mesénquima metanéfrico, que interactúa con el receptor tirosina kinasa c-Ret expresado en la yema ureteral para inducir la ramificación del conducto néfrico13. Existen varios genes candidatos: BMP4, RET, GDNF, FREM2 (FRAS1-related extracellular matrix protein 2) y FRAS1 (Fraser syndrome 1)13-16. FRAS1 y FREM2 se encargan de mantener la integridad de diversos epitelios renales, estando implicados en la iniciación del riñón metanéfrico. El gen FRAS1 codifica Fras1, una proteína que contiene repeticiones del dominio principal del proteoglicano condroitín sulfato, cuya función es mantener la integridad de la célula epitelial. La proteína Fras1 se detectó en varios tejidos en desarrollo. En el metanefros, Fras1 se detectó en la matriz extracelular recubriendo la superficie basal del uréter en crecimiento y la membrana basal del túbulo colector. En individuos con síndrome de Fraser, todas las mutaciones de FRAS1 comunicadas fueron homocigotas o heterocigotas compuestas, indicando un mecanismo genético recesivo. En contraste, en la agenesia renal unilateral no sindrómica se han observado mutaciones heterocigotas únicas de FRAS116. Por otra parte, la agenesia renal unilateral se asocia a anomalías congénitas del sistema reproductor y de la vía urinaria (quistes de las vesículas seminales)17. El 40-50 % de los niños con agenesia renal presentan otros defectos genitourinarios, incluyendo el reflujo vesicoureteral, ectopia renal, dilatación pielocalicial, duplicación ureteral, vejiga neurogénica, criptorquidia y defectos estructurales de la vagina o del útero. El 15 % de estos niños tienen defectos del sistema cardiovascular18.
La importancia de la asociación de PQRAD y agenesia renal contralateral radica, por un lado, en la posible relación causal entre ambos procesos y, por otro, en la influencia que puede tener la presencia de un riñón poliquístico único en el desarrollo precoz de IRC. De los 8 casos de PQRAD y agenesia renal unilateral (incluido el nuestro), solo en 2 se realizó estudio genético para diferenciar entre PKD1 y PKD212 y en cada caso se encontró una mutación diferente del gen PKD1. La agenesia renal unilateral, cuando ocurre como una anomalía aislada, es asintomática, suele descubrirse incidentalmente en ecografías de rutina y habitualmente se produce una hipertrofia compensadora del riñón contralateral. En el caso de la asociación con PQRAD, podría no producirse hipertrofia compensadora, acelerando el desarrollo de IRC por parte del riñón poliquístico único. Así, la PQRAD unilateral representaría un modelo para analizar las consecuencias de la reducción del número de nefronas en la PQRAD. Los casos de PQRAD y riñón único no proporcionan suficiente información sobre los volúmenes renales o las tasas de progresión8-12 y algunos ya tenían IRC avanzada a su presentación (tabla 1). Además, estos casos con PQRAD y riñón único no pueden ser comparables con los pacientes con 2 riñones poliquísticos. Los 2 pacientes de Poster12, con PQRAD unilateral, tenían volúmenes renales y tasas de incremento del volumen mayores que la media de los valores del grupo control. Con la excepción del caso de Sirvent11, que presentó hipertensión grave incontrolada, los 7 pacientes con PQRAD unilateral no tuvieron enfermedad acelerada. Nuestro paciente experimentó progresión hacia la IRC terminal, con una pérdida anual de función renal de 7,6 ml/min/1,73 m2 en los últimos 6 años. Por lo tanto, los datos existentes indican que los riñones afectados por PQRAD unilateral no parecen retener capacidad para compensar la reducción del número de nefronas. Por otro lado, la agenesia renal unilateral se presentó más frecuentemente en el lado izquierdo19, ya que en 6 de los 8 casos de PQRAD y agenesia renal contralateral el riñón ausente fue el izquierdo (75 % de los casos). Una explicación para esta diferencia en lateralidad permanece sin aclarar, ilustrando así lo limitado de nuestro conocimiento de la embriogénesis en general y de la nefrogénesis en particular.
Tabla 1. Pacientes con poliquistosis renal autosómica dominante y agenesia renal
contralateral comunicados en la literatura 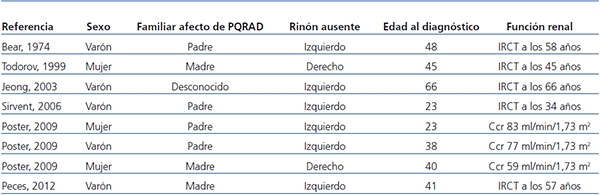
IRCT: insuficiencia renal crónica terminal; PQRAD: poliquistosis renal autosómica dominante.
En resumen, este caso ilustra la coincidencia de dos enfermedades renales hereditarias: una autosómica dominante, la PQRAD, y la otra recesiva, la agenesia renal contralateral. Aunque la PQRAD y la agenesia renal unilateral pueden ser debidas a alteraciones de 2 genes independientes, no puede excluirse la existencia de una relación causal entre ambas20. Tendrán que analizarse más casos para encontrar la respuesta a esta cuestión no resuelta.
Agradecimientos
Este estudio se llevó a cabo, en parte, con una ayuda del Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación (EC08/00236) y el Programa Intensificación Actividad Investigadora (IdiPAZ y Agencia Laín-Entralgo/CM) a R.P.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
Ramón Peces1, Cristina Vega1, Ana Aguilar1, Rosa Zometa1, Claudia Tapia1, Carlos Peces2 y Emilio Cuesta3
1Servicio de Nefrología. Hospital Universitario La Paz, IdiPAZ. Madrid
2Área de Tecnología de la Información. SESCAM. Toledo
3Servicio de Radiología. Hospital Universitario La Paz, IdiPAZ. Madrid
Referencias Bibliográficas
1. Masoumi A, Reed-Gitomer B, Kelleher C, Bekheirnia MR, Schrier RW. Developments in the management of autosomal dominant polycystic kidney disease. Ther Clin Risk Manag 2008;4:393-407. [ Links ]
2. Shiroyanagi Y, Suzuki M, Matsuno D, Mochizuki K, Kitagawa N, Tanaka M, et al. Asymmetric development of tumor-like cysts in a child with autosomal dominant polycystic kidney disease. J Pediatr Surg 2008;43:e21-3. [ Links ]
3. Wilson RD, Baird PA. Renal agenesis in British Columbia. Am J Med Genet 1985;21:153-69. [ Links ]
4. Costantini F. Renal branching morphogenesis: concepts, questions, and recent advances. Differentiation 2006;74:402-21. [ Links ]
5. Dressler GR. Advances in early kidney specification, development and patterning. Development 2009;136:3863-74. [ Links ]
6. Reidy KJ, Rosenblum ND. Cell and molecular biology of kidney development. Semin Nephrol 2009; 29:321-37. [ Links ]
7. Yosypiv IV. Renin-angiotensin system in ureteric bud branching morphogenesis: insights into the mechanisms. Pediatr Nephrol 2011;26:1499-512. [ Links ]
8. Bear RA. Solitary kidney affected with polycystic disease: A report of 2 cases. J Urol 1974;111:566-7. [ Links ]
9. Todorov VV. The diagnostic dilemma of the unilateral cystic kidney-ADPKD with aplasia of one kidney. Nephrol Dial Transplant 1999;14:2775. [ Links ]
10. Jeong GH, Park BS, Jeong TK, Ma SK, Yeum CH, Kim SW, et al. Unilateral autosomal dominant polycystic kidney disease with contralateral renal agenesis: Acase report. J Korean Med Sci 2003; 18:284-6. [ Links ]
11. Sirvent AE, Enríquez R, Ardoy F, Amorós F, González C, Reyes A. Autosomal dominant polycystic kidney disease with congenital absence of contralateral kidney. Int Urol Nephrol 2006;38:773-4. [ Links ]
12. Poster D, Kistler AD, Krauer F. Kidney function and volume progression in unilateral autosomal dominant polycystic kidney disease with contralateral renal agenesis or hypoplasia: a case series. Am J Kidney Dis 2009;54:450-8. [ Links ]
13. Song R, Yosypiv IV. Genetics of congenital anomalies of the kidney and urinary tract. Pediatr Nephrol 2011;26:353-64. [ Links ]
14. Skinner MA, Safford SD, Reeves JG, Jackson ME, Freemerman AJ. Renal aplasia in humans is associated with RET mutations. Am J Hum Genet 2008;82:344-51. [ Links ]
15. Rozen EJ, Schmidt H, Dolcet X, Basson MA, Jain S, Encinas M. Loss of sprouty1 rescues renal agenesis caused by Ret mutation. J Am Soc Nephrol 2009;20:255-9. [ Links ]
16. Saisawat P, Tasic V, Vega-Warner V, Kehinde EO, Günther B, Airik R, et al. Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Kidney Int 2012;81:196-200. [ Links ]
17. Nakanishi K, Yoshikawa N. Genetic disorders of human congenital anomalies of the kidney and urinary tract (CAKUT). Pediatr Int 2003;45:610-6. [ Links ]
18. Szmigielska A, Roszkowska-Blaim M, Werner B, Kaminska H, Brzewski M. Hypertension in a girl with severe coarctation of the aorta and renal agenesis. J Pediatr 2012;160:705-6. [ Links ]
19. Schreuder MF. Unilateral anomalies of kidney development: why is left not right? Kidney Int 2011;80:740-5. [ Links ]
20. Kerecuk L, Long DA, Ali Z, Anders C, Kolatsi-Joannou M, Scambler PJ, et al. Expression of Fraser syndrome genes in normal and polycystic murine kidneys. Pediatr Nephrol 2012; 27:991-8. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Ramón Peces
Servicio de Nefrología
Hospital Universitario La Paz, IdiPAZ, Madrid
rpeces.hulp@salud.madrid.org

