










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkNefrología (Madrid)
versão On-line ISSN 1989-2284versão impressa ISSN 0211-6995
Nefrología (Madr.) vol.33 no.5 Cantabria 2013
https://dx.doi.org/10.3265/Nefrologia.pre2013.Jun.12085
CD80, suPAR and Nephrotic Syndrome in a case of NPHS2 mutation
CD80, suPAR y síndrome nefrótico en un caso de mutación del gen NPHS2
Gabriel Cara-Fuentes1, Carlos Araya1, Changli Wei2, Christopher Rivard3, Takuji Ishimoto3, Jochen Reiser4, Richard J. Johnson3, Eduardo H. Garin1
1Division of Pediatric Nephrology, Department of Pediatrics. University of Florida. Gainesville, Florida (USA)
2Department of Medicine, Miller School of Medicine. University of Miami. Miami, Florida (USA)
3Division of Renal Diseases and Hypertension, Department of Medicine. University of Colorado. Denver, Colorado (USA)
4Department of Medicine. Rush University Medical Center. Chicago, Illinois (USA)
ABSTRACT
Background: Podocin mutations are characterized by progression to end stage renal disease and histologic findings of Focal Segmental Glomerulosclerosis (FSGS). CD80 is a podocytes protein that may play a role in proteinuria, particularly in Minimal Change Disease whereas the soluble urokinase receptor (suPAR) is characteristically elevated in the serum of FSGS patients.
Methods: In a patient with nephrotic syndrome and podocin mutation, urinary and serum CD80 as well as suPAR were measured using commercially available kits. Urinary CD80 molecular size was determined by western blot analysis. Glomerular staining for CD80 and podocin was performed.
Results: Patient displayed marked elevated CD80 and mildly increased suPAR urinary levels compared to controls. Serum CD80 level was within the range observed in normal controls. Serum suPAR level was elevated, albeit in the lower range reported for patients with primary FSGS. Immunofluorescence examination of kidney biopsy revealed glomerular CD80 expression.
Conclusion: The combination of serum and urinary biomarkers can help differentiate various forms of FSGS. High urinary CD80 and elevated serum and urinary suPAR might represent a profile to differentiate this genetic form of FSGS from primary FSGS.
Key Words: CD80, Nephrotic syndrome, Podocyte, Podocin mutation, suPAR.
RESUMEN
Antecedentes: Las mutaciones de la podocina están caracterizadas por la progresión hacia enfermedad renal terminal y por hallazgos histológicos de glomeruloesclerosis segmentaria y focal (GSF). CD80 es una proteína podocitaria que parece tener un papel en la proteinuria de la enfermedad de cambios mínimos, mientras que el receptor soluble de la uroquinasa (suPAR) es característicamente elevado en el suero de pacientes con GSF.
Métodos: En un paciente con síndrome nefrótico y mutación de la podocina, se cuantificó CD80 y suPAR en suero y orina usando los kits disponibles en el mercado. El peso molecular del CD80 urinario fue determinado mediante Western blot. Se realizó la tinción para CD80 y podocina en el glomérulo.
Resultados: El paciente presentó niveles urinarios marcadamente elevados de CD80 y ligeramente elevados de suPAR en comparación con controles. El nivel sérico de CD80 se encontró dentro del rango observado en controles. El nivel sérico de suPAR fue elevado, aunque en el límite inferior del rango publicado para pacientes con GSF primaria. La inmunofluorescencia de la biopsia renal mostró expresión glomerular de CD80.
Conclusión: La combinación de biomarcadores séricos y urinarios quizás ayude a diferenciar entre diferentes formas de GSF. Niveles elevados de CD80 en orina y suPAR en suero quizás representen un perfil característico que permita diferenciar entre esta forma genética de GSF y GSF de causa primaria.
Palabras clave: CD80, Síndrome nefrótico, Podocito, Mutación de la podocina, suPAR.
Introduction
Podocytes are specialized visceral epithelial cells that constitute an integral part of the glomerular capillary wall.1 Podocyte foot processes (FP) are dynamic structures formed by an actin-myosin based contractile cytoskeleton that are connected to the glomerular basement membrane (GBM) via transmembrane receptors including integrins.2 The FP of neighbouring podocytes regularly interdigitate with each other, leaving between them filtration slits that are bridged by modified adherens junctions, referred to as the slit diaphragm (SD).
The SD is a multiprotein complex composed of a growing number of constituents including nephrin (NPHS1), podocin (NPHS2), TRPC6, PLCE1, α-actinin-4, CD2AP, P-cadherin, ZO-1, FAT and Neph 1-3.3 These proteins are directly or functionally connected to the actin-based cytoskeleton. The maintenance of the podocyte's architecture as well as signaling pathways between cytoskeleton, FPs and the SD are critical for the integrity of the podocyte, and hence, the integrity of the glomerular barrier.
Studies in both experimental and some human glomerular diseases suggest that proteinuria is due to a direct defect within podocytes or due to abnormal extracellular signalling involving the podocyte.4-6 Under specific experimental conditions, such as glomerular injury induced by lipopolysaccharide (LPS) and puromycin aminonucleside,4 podocytes express CD80, a transmembrane protein that has dual specificity for two CD28 family members, the stimulatory receptor CD28 and the inhibitory receptor CTLA-4 (CD152).7 Alternatively, podocytes upscale the expression of urokinase plasminogen activator receptor uPAR,8 a three-domain-glycosylphosphatidylinositol-anchored protein which acts not only as a proteinase receptor but can also regulate cell migration, adhesion, differentiation and proliferation through integrin binding.9 The LPS mouse model has been useful to define the role of both podocyte CD80 and uPAR in the development of proteinuria. Specifically, proteinuria is not observed in either the CD80 or uPAR (Plaur -/-) knockout mice following LPS administration.4,8
The role of CD80 and uPAR in human glomerular disease is still being unravelled. Subjects with Minimal Change Disease (MCD) show high urinary CD80 excretion associated with high podocyte CD80 expression.5 Urinary CD80 is not elevated in subjects with idiopathic FSGS.6 In contrast, patients with primary Focal Segmental Glomerulosclerosis (FSGS) have elevated expression of uPAR in podocytes8 as well as high serum levels of suPAR. Both forms of uPAR bind to the podocyte surface to activate integrins.6
Podocin (NPHS2) mutations represent the most common cause of genetic nephrotic syndrome (NS) in humans. Subjects with these mutations typically present with steroid resistant nephrotic syndrome that frequently progresses to end-stage-renal disease.11 Here we report a patient with steroid-resistant nephrotic syndrome secondary to an NPHS2 mutation. In this patient the renal biopsy showed CD80 expression in glomeruli which was associated with marked elevated urinary levels of CD80 and increased serum suPAR level.
Case report
A 5 year-old Caucasian boy was referred to our hospital with a 12-month history of intermittent periorbital and pretibial edema. His past medical history was significant for congenital hypothyroidism, cerebral palsy with right hemiparesis, mild development delay, facial dimorphism and mild, intermittent asthma.
Physical examination was remarkable for anasarca. Blood pressure was 102/52mmHg. Thyroid was not enlarged. Urinalysis revealed protein >300mg/dL and 1 RBC/HPF. Random urine protein/creatinine ratio was 16. The boy demonstrated hypoalbuminemia (1.5g/dL), hypercholesterolemia (534mg/dL), with a normal serum creatinine 0.3mg/dL. Liver function tests, C3 and C4 were normal. Thyroid function test revealed a lower free T4 (0.7ng/dL), elevated TSH (10.7mIU/L), with negative thyroid peroxidase and thyroglobulin antibodies.
Because of the age of the patient at presentation and the clinical and laboratory findings, a presumed diagnosis of minimal change disease was made and steroid therapy was started at 2mg/kg/day. Anasarca and massive proteinuria (urine protein/creatinine ratio of 28) persisted despite a 4 week-course of steroid treatment. The patient was considered steroid resistant and a renal biopsy was performed. Histopathologic examination of the kidney biopsy showed mild mesangial expansion and cellularity, mild interstitial fibrosis and foam cells. Electron microscopy did not show electron dense deposits.
Steroid treatment was tapered and tacrolimus was started at a dose of 0.1mg/kg/day. In spite of 6 months of tacrolimus therapy, his nephrotic syndrome persisted. At this time, a genetic analysis for nephrin and podocin was requested. The test results revealed the presence of a mutation in the NPHS2 gene (see below).
Tacrolimus was discontinued. Edema was treated with diuretics (furosemide, spironolactone, metolazone), proteinuria with the use of enalapril, and hypercholesterolemia by simvastatin. After 3.5 years of follow-up, he has persistent nephrotic syndrome (on last evaluation, mild edema, albumin 1.8g/dL, cholesterol 449mg/dL, protein/creatinine ratio of 28.5), serum creatinine has increased to 6.2mg/dL (- GFR 10mL/min/1.73m2-) and is currently undergoing hemodialysis.
Methods
The study was approved by the Institutional Review Board of the University of Florida.
Urinary CD80 and uPAR were measured using commercially available kits (Bender MedSystems, Burlingame, CA and R&D Systems, respectively).
CD80 urinary Molecular Weight (MW) (Figure 1). Frozen (-80 C) urine samples were warmed to room temperature, vortexed, and centrifuged at 8.000xg for 5 min. Urine creatinine was determined using the ACE autoanalyzer (Alfa Wasserman, West Caulfield, NJ). Proteins were loaded for an equivalent amount (7.5ug) of urinary creatinine per lane in a 4-20% Criterion polyacrylamide gel (Bio-Rad, Hercules, CA) and separated at 120V for 90 minutes. Proteins were transferred to PVDF membrane, blocked for 1 hour in 5% milk and incubated overnight at 4 C with anti-CD80 antibody (R&D System, AF140, Minneapolis, MN). The membrane was washed in TTBS and revealed using an anti mouse HRP linked secondary antibody (Santa Cruz Biotech, SC-2354, Santa Cruz, CA) and Immun-Star reagent (Bio-Rad). The membrane was exposed to film and images documented with an Epson 4990 scanner (LongBeach, CA).

Figure 1. Western blot of CD80 protein in urine
From a patient with nephrotic syndrome due to podocin mutation (a) and control subject (b).
Glomerular staining for CD80 and podocin was performed as previously described.6
Results
Genetic analysis. DNA analysis of NPHS2 gene showed a homozygous mutation with G to A substitution in nucleotide position 413, codon position 138, resulting in an amino acid change from arginine to glutamine (p.R138Q). This is a known recessive mutation that causes steroid resistant nephrotic syndrome.12,13
Serum and urinary CD80 and suPAR levels (Table 1). The patient showed marked elevated CD80 urinary levels similar to those seen in MCD and urinary suPAR levels were also elevated when compared to controls. Serum CD80 was within the range observed in normal controls and in patients with MCD and FSGS. Serum suPAR was below the mean reported in patients with FSGS.
Table 1. 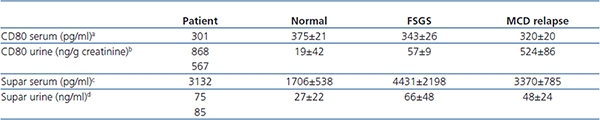
aReference: 5. bReference: 10. cReference: 6. dReference: unpublished data.
Western blot analysis for CD80 protein in urine is shown in Figure 1. Urinary CD80 protein was present in our patient and absent in urine of a normal control. Its MW is 55kDa.
Immunofluorescence examination of a biopsy revealed CD80 (red stain) in the glomeruli of our patient with nephrotic syndrome secondary to NPSH2 mutation in relapse (Figure 2 A), podocin (green stain, Figure 2 B), and CD80 co-localization with podocin along the glomerular capillary wall (Figure 2 C). The CD80 and podocin glomerular staining in this patient was similar to the one observed in glomeruli of MCD patients in relapse (Figure 2 D, Figure 2 E, Figure 2 F).

Figure 2. CD80 and podocin
CD80 (red stain) and podocin (green stain) in glomerulus of (a-b) genetic nephrotic syndrome (podocin mutation)
and (d-e) minimal change disease in relapse. Combinated CD80 and podocin is expressed in glomeruli from
a patient with podocin mutation (c) and a MCD patient in relapse (f).
Discussion
Podocin is a hairpin-like protein of 383 amino acids located at the podocyte cell membrane facing the slit diaphragm with both N-terminal and C-terminal domains ending in the cytoplasm.2,14 Podocin is encoded by the NPHS2 gene mapped to chromosome 1q25-q31.
More than 50 NPHS2 mutations have been reported that occurred as homozygous or compound. Heterozygous R138Q was most frequently found in Germany and France while the P20L variant was observed mainly in Italy.15,16
Mutations of the NPHS2 gene cause 10-28% of all non-familial childhood steroid resistant nephrotic syndrome (SRNS).17 There is a strong correlation between causative gene mutations and the age of onset of the nephrotic syndrome. The presence of at least one truncating mutation, "R138Q", leads to early onset of SRNS at a median age of 1.7 years rather than 4.7 years.18 Compound heterozygosity for the R229Q variant of podocin and one "bonafide" podocin mutation causes adult onset in up to 15% of SRNS cases.19
Our patient has the p.R138Q mutation and his nephrotic syndrome presented at less than 5 years of age. His glomerular pathological findings were consistent with those observed in early stages of the nephrotic syndrome in patients with podocin mutation. Nephrotic syndrome caused by podocin mutation is considered a podocytopathy. Podocytopathy has been defined as a spectrum of proteinuric glomerular diseases resulting from podocyte abnormalities.20
Given the ability of podocyte CD80 to sequester slit diaphragm proteins and thus disturb slit diaphragm function4 we analyzed glomerular CD80 expression in our patient. Our patient had evidence for both glomerular expression of CD80 and increased urinary CD80 levels. The molecular weight of CD80 in the urine was determined to be approximately 55kDa and is thus consistent with cell-membrane bound CD80. In comparison, shedding of the extracellular domain portion of CD80 results in detection of a cleaved CD80 protein fragment reduced in MW.
The role of CD80 in proteinuria in this patient is not known. The mutation in podocin itself is expected to alter slit diaphragm function. Podocin is associated with specialized lipid raft microdomains of the podocyte plasma membrane and recruits nephrin into rafts. In contrast, disease-causing mutations of podocin (R138Q and R138X) failed to recruit nephrin into rafts either because these mutants were retained in the endoplasmic reticulum (R138Q), or because they failed to associate with rafts (R138X) despite their presence in the plasma membrane.21 Conversely, proteinuria could result of changes in the binding properties to essential slit diaphragm proteins.22 Dysfunctional podocin might also explain the release of CD80 into the urine as perhaps the CD80 was induced by the podocyte as a consequence of podocyte activation from the genetic mutation similar to CD80 induction after deletion of α3 integrin.4
In patients with primary FSGS, serum suPAR level is increased.6 suPAR has been shown to increase glomerular permeability and cause proteinuria through binding to and activation of podocyte β3 integrins.6 The level of circulating uPAR (suPAR) in our patient was elevated and within the range of levels observed in patients with FSGS as per recently published reference.6 Our patient had also an increased level of urinary suPAR when compared to controls. This is consistent with the notion that suPAR is filtered by the glomerulus under normal conditions and the increase in urinary level of suPAR is likely due to the enhanced suPAR serum level seen in this patient. It is interesting and will require further studies as to why suPAR is induced in this patient with genetic FSGS. In contrast with FSGS patients, suPAR levels in MCD patients are generally not different than those seen in normal controls.6 Therefore, the elevated suPAR levels in the relapsing MCD cases reported in this paper, might be indicative of poor prognosis and might help identify cases of Minimal Change Disease that are transitioning into FSGS.
In summary, the hybrid profile of a MCD marker such as podocyte CD80 together with the excretion of urinary CD80 together with higher levels of the FSGS marker suPAR in blood and urine might be characteristic for NPHS2 mutations and contribute to the proteinuria that occurs in these patients. The observation that the podocin mutation can be associated with high urinary CD80 suggests it is different from idiopathic FSGS (in which urinary CD80 is low) and is more similar to that which is observed with MCD. High urinary CD80, elevated serum and urinary suPAR might represent a profile to differentiate this genetic form of FSGS from primary FSGS. This report emphasizes the importance of more future studies that include biomarkers in blood and urine of distinct glomerular diseases in combination with renal biopsy that can be subject to sampling error. Such an approach will allow to achieve a refined diagnosis of the underlying disease process and might be used to distinguish milder from aggressive forms of glomerular disease.
Conflicts of interest
The authors declare that they have no conflicts of interest related to the contents of this article.
References
1. Henique C, Tharaux PL. Targeting signaling pathways in glomerular diseases. Curr Opin Nephrol Hypertens 2012;21:417-27. [ Links ]
2. Ruoslahti E. Integrins. J Clin Invest 1991;87:1-5. [ Links ]
3. Patrakka J, Tryggvason K. Molecular make-up of the glomerular filtration barrier. Biochem Biophys Res Commun 2010;396:164-9. [ Links ]
4. Reiser J, von Gersdorff G, Loos M, Oh J, Asanuma K, Giardiano L, et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest 2004;113:1390-7. [ Links ]
5. Garin EH, Diaz LN, Mu W, Wasserfall C, Araya C, Segal M, et al. Urinary CD80 excretion increases in idiopathic minimal-change disease. J Am Soc Nephrol 2009;20:260-6. [ Links ]
6. Wei C, El Hindi S, Li J, Fornoni A, Goes N, Sageshima J, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med 2011;17:952-60. [ Links ]
7. Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol 2005;23:515-48. [ Links ]
8. Wei C, Möller CC, Altintas MM, Li J, Schwarz K, Zacchigna S, et al. Modification of kidney barrier function by the urokinase receptor. Nat Med 2008;14:55-63. [ Links ]
9. Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol 2002;3:932-43. [ Links ]
10. Garin EH, Mu W, Arthur JM, Rivard CJ, Araya CE, Shimada M, et al. Urinary CD80 is elevated in minimal change disease but not in focal segmental glomerulosclerosis. Kidney Int 2010;78:296-302. [ Links ]
11. Ruf RG, Lichtenberger A, Karle SM, Haas JP, Anacleto FE, Schultheiss M,et al. Arbeitsgemeinschaft Für Pädiatrische Nephrologie Study Group. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J Am Soc Nephrol 2004;15:722-32. [ Links ]
12. Tonna SJ, Needham A, Polu K, Uscinski A, Appel GB, Falk RJ, et al. NPHS2 variation in focal and segmental glomerulosclerosis. BMC Nephrol 2008;9:13. [ Links ]
13. Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 2000;24:349-54. [ Links ]
14. Roselli S, Gribouval O, Boute N, Sich M, Benessy F, Attié T, et al. Podocin localizes in the kidney to the slit diaphragm area. Am J Pathol 2002;160:131-9. [ Links ]
15. Franceschini N, North KE, Kopp JB, McKenzie L, Winkler C. NPHS2 gene, nephrotic syndrome and focal segmental glomerulosclerosis: a HuGE review. Genet Med 2006;8:63-75. [ Links ]
16. Caridi G, Perfumo F, Ghiggeri GM. NPHS2 (Podocin) mutations in nephrotic syndrome. Clinical spectrum and fine mechanisms. Pediatr Res 2005;57:54R-61R. [ Links ]
17. Chernin G, Heeringa SF, Gbadegesin R, Liu J, Hinkes BG, Vlangos CN, et al. Low prevalence of NPHS2 mutations in African American children with steroid-resistant nephrotic syndrome. Pediatr Nephrol 2008;23:1455-60. [ Links ]
18. Hinkes B, Vlangos C, Heeringa S, Mucha B, Gbadegesin R, Liu J, et al. APN Study Group. Specific podocin mutations correlate with age of onset in steroid-resistant nephrotic syndrome. J Am Soc Nephrol 2008;19:365-71. [ Links ]
19. Machuca E, Hummel A, Nevo F, Dantal J, Martinez F, Al-Sabban E, et al. Clinical and epidemiological assessment of steroid-resistant nephrotic syndrome associated with the NPHS2 R229Q variant. Kidney Int 2009;75:727-35. [ Links ]
20. Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int 2007;71:1205-14. [ Links ]
21. Huber TB, Simons M, Hartleben B, Sernetz L, Schmidts M, Gundlach E, et al. Molecular basis of the functional podocin-nephrin complex: mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft microdomains. Hum Mol Genet 2003;12:3397-405. [ Links ]
22. Reiser J, Polu KR, Möller CC, Kenlan P, Altintas MM, Wei C, et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet 2005;37:739-44. [ Links ]
![]() Correspondence:
Correspondence:
Eduardo H. Garin,
Division of Pediatric Nephrology,
Department of Pediatrics,
University of Florida,
1600 SW Archer Rd.
HD214. Gainesville, Fl,
32610, Gainesville, Florida, USA
garineh@peds.ufl.edu
Enviado a Revisar: 20 Ene. 2013
Aceptado el: 16 Jun. 2013

