










 Serviços customizados
Serviços customizados
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkIntroducción
Los feocromocitomas (PH) y paragangliomas (PGL) son tumores neuroendocrinos productores de catecolaminas derivados de las células cromafines de la cresta neural. La mayoría aparecen en la médula adrenal (PH), pero el 20% son extraadrenales1.
Los PH/PGL son una causa rara de hipertensión arterial secundaria, con una incidencia en pacientes hipertensos comprendida entre el 0,3 y el 0,5%. La presentación típica es la combinación de un grado variable de hipertensión arterial acompañada de sintomatología paroxística, espontánea o inducida por un aumento de la presión intraabdominal u otros estímulos. Paradójicamente, en algunos pacientes con PH/PGL, pueden aparecer episodios de hipotensión ortostática e incluso síncope, que se deben a la desensibilización del receptor vasomotor o a la disminución de volumen intravascular2. En algunas ocasiones, las manifestaciones clínicas de estos tumores no tienen relación con cambios en las cifras tensionales; así que, en algunos casos, predominan síntomas de otros tumores que se asocian con PH/PGL, en el contexto determinadas mutaciones genéticas (cáncer medular de tiroides, cáncer de células renales, hemangioblastomas, neurofibromas, tumores pancreáticos neuroendocrinos, tumor del estroma gastrointestinal (GIST), tumores hipofisarios…)3.
En los tumores localizados en la cabeza y el cuello, no se producen estos síntomas comentados, y se suelen presentar como una masa de tamaño creciente no productora de catecolaminas1,2.
Según la serie analizada, hasta el 35% de los casos de PH y el 15% de los PGL se asocian a una mutación germinal, por lo que se pueden considerar tumores neuroendocrinos genéticamente determinados4-6. Hoy en día se conocen hasta 19 genes relacionados con la fisiopatogenia de estos tumores (tabla 1)2,3,5,7.
Tabla 1 Principales características en la presentación clínica, fenotipo bioquímico de los feocromocitomas/paragangliomas y sus mutaciones genéticas

A: adrenalina; DA: dopamina; GITS: tumores del estroma gastrointestinal; N/D: no determinado; NA: noradrenalina; SNC: sistema nervioso central.
Fuente: Adaptado de Pacak y Wimalawansa5, Jafri y Maher7, Burnichon et al.37 y Reisch et al.38.
Sin embargo, de todos estos genes, los implicados con más frecuencia en las mutaciones germinales en la práctica clínica son los genes von Hippel Lindau (VHL) y el complejo succinato deshidrogenasa (SDHx) D, B y C4,5,8.
Los estudios de expresión génica han objetivado posibles mecanismos fisiopatológicos comunes para algunos de estos genes, clasificándolos en 2 grupos o «clusters» principales, según las vías de transcripción implicadas.
El primer «cluster» se relaciona con la activación de las vías de la angiogénesis y pseudohipoxia, y comprende las mutaciones en el gen de VHL y SDHx. El segundo «cluster» lo componen los tumores asociados con la mutación del gen de la neurofibromatosis tipo 1 (NF1) y del protooncogén RET (neoplasia endocrina múltiple tipo 2 o MEN2), que producen una alteración en la regulación del metabolismo adrenérgico, síntesis de proteínas y en la vía de señalización de la cinasa6,7. Estos perfiles de expresión genética se pueden utilizar para clasificar los PH/PGL, asignándolos a uno de los 2 grupos, cada uno de los cuales tendría dianas terapéuticas específicas4,8.
Para poder identificar correctamente una mutación genética y, por tanto, realizar un diagnóstico diferencial genético adecuado, el clínico debe realizar una buena historia clínica personal y familiar, y tener en cuenta los siguientes aspectos principales: la edad de presentación, la localización del tumor, el fenotipo bioquímico y la presencia de otros tumores asociados5.
Aproximación tradicional al diagnóstico de los PH/PGL
Diagnóstico bioquímico: conceptos básicos
Las principales indicaciones para iniciar un estudio bioquímico de evaluación de un posible PH/PGL son: la sospecha clínica (hipertensión arterial paroxística, cefalea, diaforesis, palpitaciones), una inexplicable variabilidad en la tensión arterial, crisis hipertensivas ante ciertos estímulos (inducción anestésica, fármacos, contrastes radiológicos, cirugía, ejercicio, parto), incidentalomas suprarrenales y antecedentes familiares de PH/PGL. En pacientes jóvenes con hipertensión y peso corporal normal, la presencia de diabetes también es un indicio clínico para la sospecha de PH9,10.
Los PH y PGL sintetizan, almacenan y secretan catecolaminas: dopamina (DA), adrenalina (A) y noradrenalina (NA). Las catecolaminas son parcial o totalmente convertidas en metabolitos inactivos, metanefrina y normetanefrina, por la catecol-O-metiltransferasa presente en las células tumorales11,12.
La liberación de catecolaminas al plasma puede ser paroxística o mínima, mientras que la liberación de metanefrinas es más sostenida en el tiempo, ya que el metabolismo intratumoral de las catecolaminas se lleva a cabo independientemente de la liberación de las mismas y, por tanto, su liberación al plasma tiene lugar casi siempre de una forma continuada5. Es por ello por lo que la determinación de metanefrinas en plasma y orina es más sensible que la de catecolaminas, con menor número de falsos negativos. En otras palabras, unos niveles normales de metanefrinas en plasma u orina excluyen con bastante fiabilidad la presencia de un tumor productor de catecolaminas, excepto en los tumores pequeños (<1 cm), en los que no liberan catecolaminas, y en los excepcionales casos de tumores exclusivamente productores de DA5,11.
En los pacientes con elevaciones leves de catecolaminas o metanefrinas es importante descartar la presencia de falsos positivos, y tener en cuenta que existen fármacos que aumentan la liberación de catecolaminas. Además, situaciones como los eventos cardiovasculares agudos pueden elevar también las catecolaminas de forma transitoria (tabla 2)2,13. Los pacientes con enfermedad renal crónica, sobre todo aquellos en diálisis, tienen frecuentemente niveles elevados de metanefrinas en plasma en ausencia de PH/PGL, por lo que hay que tener especial precaución en la interpretación de los resultados bioquímicos en esta población14.
Tabla 2 Causas que pueden aumentar las concentraciones de catecolaminas (falsos positivos)

ACVA: accidente cerebrovascular agudo; IAM: infarto agudo de miocardio; IMAO: inhibidor de la mono-amino oxidasa.
Fuente: Adaptado de Kronenberg et al.39.
Los pacientes con falsos positivos suelen tener mayores incrementos de catecolaminas en comparación con los niveles de metanefrinas, debido a la activación simpatoadrenal, de forma que un ratio de normetanefrina/noradrenalina (NA)<0,52 y de metanefrina/adrenalina (A)<4,2 señala una activación simpatoadrenal11. La prueba de supresión con clonidina puede ser un método para distinguir los verdaderos de los falsos positivos con niveles límite de NA. La clonidina es un agonista de los receptores α2-adrenérgicos, que inhibe la liberación de NA neuronal en pacientes sin PH/PGL, pero no ante la presencia de una secreción tumoral autónoma de catecolaminas. Esta prueba tiene una especificidad de un 100%, con una sensibilidad del 97%, pero hasta ahora no ha sido validada en ningún estudio prospectivo2,5.
Para discriminar las diferentes formas hereditarias es importante tener en cuenta que la NA se convierte en A mediante la feniletanolamina-N-metiltransferasa (PNMT) solo en las células cromafines de la médula adrenal. Así pues, la A se produce casi de forma exclusiva en los tumores adrenales, mientras que los tumores extraadrenales producen principalmente NA. Este es debido a que el cortisol es capaz de inducir la síntesis de PNMT a nivel adrenal y a que el acúmulo de succinato en el tejido tumoral produce una hipermetilación del gen promotor de la PNMT e impide su síntesis4.
Las concentraciones plasmática y urinaria de metanefrinas pueden ser normales en algunos casos de PGL de gran tamaño, localización abdominal extraadrenal, metastásicos o con la mutación de SDHB (suelen ser células tumorales que han perdido su capacidad de síntesis por desdiferenciación)15. Esto se puede deber a un defecto en la síntesis de catecolaminas por la ausencia de la tirosina hidroxilasa (TH), una enzima limitante de la velocidad en la síntesis de catecolaminas16.
Los tumores productores únicamente de DA son raros y suelen ser PGL extraadrenales, por lo que estaría indicada su determinación o la de su metabolito (metoxitiramina) en pacientes con una presentación atípica, en los cuales la sospecha de PGL sea alta y los niveles de metanefrinas, normales11,16.
Por otra parte, los PGL de cabeza y cuello -que suelen ser bioquímicamente silentes- también pueden ser positivos para TH, que convierte la tirosina en dihidroxifenilalanina (precursor de la dopamina), de forma que hasta en el 23% de los casos pueden presentar un aumento de la excreción urinaria de 3-metoxitiramina, y en dichos tumores puede considerarse un mejor marcador que la detección de DA en orina17.
Localización: técnicas de imagen
Una vez que se ha realizado el diagnóstico bioquímico, hay que localizar el tumor. La tomografía computarizada o la resonancia magnética nuclear son las pruebas de imagen iniciales en el diagnóstico; estos tumores presentan unas características propias18,19. Hay que tener en cuenta que hasta el 25% de todos los PH se descubren de manera incidental y, según las series, hasta el 8% de los incidentalomas suprarrenales son PH7,20.
Sin embargo, si el tumor es de pequeño tamaño, se encuentra en una localización inusual o hay cambios posquirúrgicos, estas pruebas pueden no detectarlo.
Por tanto, se recomienda complementar el diagnóstico anatómico con una prueba de imagen funcional, sobre todo con la sospecha de tumores extraadrenales, síndrome genético (fenotipo característico o edad joven), gran tamaño (>3 cm) o bilateralidad21.
Las más utilizadas son la gammagrafía con metaiodobenzilguanidina (MIBG) y la PET con 18F-fluorodopamina (18F-FDA) o con 18F-fluorodihidroxifenilalanina (18F-FDOPA).
Las pruebas funcionales de imagen se basan en el transporte de radiofármacos a través de los sistemas de transporte de catecolaminas localizados en la membrana de la célula; el más específico es el transportador de membrana de NA (NET), que se encarga de la recaptación de NA/DA.
La MIBG es un análogo de guanidina de estructura similar a la NA, el cual es captado a través del transportador NET y concentrado por los tejidos cromafines funcionantes. Además, los PH/PGL pueden captar y descarboxilar aminoácidos como la dihidroxifenilalanina (DOPA). De este modo, la 18F-FDOPA, que es un precursor de catecolaminas, es captada por el transportador de aminoácidos (LAT) de las células cromafines y convertida a 18F-FDA en estas células21 mediante la enzima descarboxilasa de aminoácidos L-aromáticos, la cual cataliza una etapa limitante en la síntesis de catecolaminas. La 18F-FDA, que es similar a la DA, es captada por las células cromafines mediante el transportador NET, pero con una mayor afinidad que la MIBG, por lo que la PET 18F-FDA tiene la mayor sensibilidad y especificidad en pacientes con diagnóstico bioquímico de PGL, cualquiera que sea su estado genético14. De forma general, la 18F-FDA o 18F-FDOPA pueden ser de elección para localizar el tumor primario, detectar enfermedad multifocal o metastásica16. El principal problema de estas técnicas es que solo están disponibles en algunos centros16,22.
En general, la MIBG tiene una sensibilidad del 80% y una especificidad del 99%. Sin embargo, la sensibilidad de 123I-MIBG desciende hasta un 56-70% en el caso de los PGL. Por tanto, no es la mejor opción para el estudio de lesiones extraadrenales, metástasis, recurrencia y en la sospecha de síndromes hereditarios (especialmente en el caso de mutaciones en los genes SDHx -sobre todo en el caso de mutaciones en SDHB- y VHL). Tampoco es una prueba óptima para el estudio de PGL en cabeza y cuello14. No obstante, en la sospecha de un PH esporádico no metastásico, la 123I -MIBG puede ser tan sensible como la PET y superior a 111In-DTPA-pentetreotida (octreoscan) en la localización del tumor21.
Además, hay que tener en cuenta otras causas de falsos negativos con MIBG, como son las lesiones pequeñas, la necrosis, los tumores poco diferenciados (pérdida de NET), los fármacos que actúen sobre el sistema adrenérgico y una intensa captación suprarrenal. De esta forma, si los niveles de metanefrinas permanecen elevados después de la cirugía de un PH se debe valorar realizar un rastreo con 123I-MIBG para descartar la presencia de metástasis, puesto que, debido a una mayor actividad metabólica del tumor primario, la primera gammagrafía puede no detectar las metástasis de pequeño tamaño21-24.
Para los casos en los que se sospeche malignidad, multifocalidad o la presencia de mutaciones germinales tipo SDHx, se recomienda el estudio funcional con otros trazadores diferentes de la 123I -MIBG. La sensibilidad de estos trazadores (18F-FDA, 18F-FDOPA) oscila desde un 81 hasta un 100%14,21,22.
Si el estudio de la PET es también negativo, probablemente nos encontremos ante un tipo inusual de PH -en el que las células tumorales no expresan el sistema transportador de NET o puede tener un número bajo de gránulos de almacenamiento de catecolaminas-, o bien un PH maligno. Entonces se puede indicar una prueba con ligandos inespecíficos, como los relacionados con el receptor de somatostatina (SSTR). En estos casos, la gammagrafía con 111In-pentetreotida puede detectar lesiones que no captan 123I-MIBG ni 18F-FDA3,22. El 111In-pentetreotida se une específicamente a los receptores de SSTR que se expresan en las membranas celulares, especialmente los subtipos 2 y 5. Los SSTR se expresan en muchas células de origen neuroendocrino y, por tanto, en los tumores derivados de ellas. Sin embargo, la expresión de SSTR puede variar y afectar a la sensibilidad de la técnica21.
Aproximación al diagnóstico desde el punto de vista genético
La historia familiar: modelos generales de herencia
Hasta el 35-45% de los casos pueden tener una mutación germinal subyacente3,6,7. Muchos de los genes implicados en PH/PGL poseen una penetrancia incompleta, o expresión fenotípica variable, y por ello aproximadamente solo el 10% de los PH/PGL tienen antecedentes familiares de PH.
La mayoría de los PH/PGL hereditarios se transmiten de forma autosómica dominante mediante un mecanismo de «second hit» (pérdida del alelo salvaje en el tumor), pero en el caso de los PH/PGL debidos a mutaciones del protooncogén RET y del factor 2A inducible por hipoxia (HIF2A), solo precisan de la mutación en un alelo para activarse (sin necesidad de «second hit»)4,5,8. Habitualmente los pacientes con PH/PGL asociados a mutaciones en los genes RET, VHL y NF1 tienen una historia familiar característica. Sin embargo, hay que tener en cuenta que aproximadamente hasta el 50% de los casos con el síndrome MEN2B o con cambios en el gen NF1 tienen una mutación de novo. Por otro lado, los pacientes con mutaciones en SDHx se presentan con historia familiar solo en el 30% de los casos, debido a la penetrancia incompleta de estos síndromes, por lo que una historia familiar negativa no excluye la presencia de un tumor hereditario. A este respecto, hay que considerar que genes como SDHD, SDHAF2 y el factor X asociado a MYC (MAX) tienen una impronta materna, es decir, solo la transmisión paterna produce la enfermedad4,5.
En niños, hasta en un 70% de los casos esporádicos se identifica una mutación germinal subyacente3. Si no hay un fenotipo característico o una historia familiar de MEN2, el diagnóstico más probable es una mutación en VHL, a la que sigue en probabilidad la mutación de SDHx9,25. Además, en la población pediátrica existe una mayor probabilidad de tumores extraadrenales, mayor tamaño, existencia de tumores sincrónicos y malignidad26.
Diagnóstico genético y fenotipo bioquímico
Se puede orientar el diagnóstico genético (tabla 1) mediante el fenotipo bioquímico de PH/PHL, que comprendería las siguientes categorías: tumores adrenérgicos (A, metanefrinametanefrina), noradrenérgicos (NA, normetanefrina), mixtos y dopaminérgicos (DA, metoxitiramina). En el caso de los mixtos, y para facilitar su diagnóstico, aquellos productores de NA y A se consideran adrenérgicos, mientras que los productores de NA y DA serían dopaminérgicos5.
Los tumores VHL casi siempre están localizados en la glándula adrenal (PH), tienen un fenotipo productor de NA y normetanefrina, y tan solo un 3% tienen elevaciones de A o metanefrina27.
En los casos de PH que producen exclusivamente NA, es improbable la presencia de síndrome MEN2 (ya que en ese caso son productores de A y metanefrina)11,28. Esto se debe a que los PH de los pacientes con MEN2 (a diferencia de VHL) expresan PNMT y tienen una mayor actividad de la TH27.
Los tumores cromafines por mutaciones en SDHx producen en su gran mayoría NA/normetanefrina28. La elevación de DA, y su metabolito metoxitiramina, también se observa en algunos casos de SDHx5,11-13,29. La metoxitiramina puede ser el único marcador en casos de tumores productores exclusivamente de DA, que suelen ser clínicamente silentes, además de ser un marcador predictor de malignidad11-13. El tejido metastásico carece de enzimas para la síntesis de catecolaminas, por lo que niveles elevados de DA, DOPA y metoxitiramina podrían ser un marcador de enfermedad metastásica11. Incluso el aumento de los niveles de metoxitiramina en plasma pueden ser un marcador más sensible de producción tumoral de DA y de diseminación metastásica que los niveles plasmáticos o urinarios de DA30.
Los genes RET, de la proteína transmembrana 127 (TMEM127), y NF1 expresan un fenotipo de secreción de A/metanefrina, mientras que las mutaciones en MAX y HIF2A expresan un fenotipo noradrenérgico4.
Para mejorar los registros genéticos y precisar mejor la incidencia y prevalencia de estos síndromes, se debería incluir a los pacientes en registros poblaciones. En nuestro país, el centro de referencia para el estudio genético de estos tumores está coordinado por la Dra. Mercedes Robledo (mrobledo@cnio.es), en el Centro Nacional de Investigaciones Oncológicas (CNIO) en Madrid.
Genética e imagen funcional
En el caso de las mutaciones de VHL, NF1 o RET, se prefiere el uso de 18F-FDA o 18F-FDOPA. En el caso de VHL, los PH suelen ser bilaterales hasta en un 50% y la 18F-FDA es superior a la MIBG debido a la poca expresión de NET en estos casos22,31,32.
Si se sospechan mutaciones SDHx, se recomienda el uso de 18F-FDA (18F -FDOPA en el caso de PGL de cabeza y cuello), 18F-FDG o 111In-DTPA-pentetreotida.
En general, los tumores asociados con determinadas mutaciones SDHx (SDHC, SDHD y SDHAF2) se encuentran en cabeza y cuello. Si se objetivasen niveles altos de catecolaminas se deberían realizar estudios de imagen para identificar otro tumor -por lo general localizado en el abdomen o pelvis-33. Las mutaciones SDHB se asocian con una mayor frecuencia a malignidad (70%) y a tumores extraadrenales. El PH puede aparecer en un 25% de los casos y el riesgo de bilateralidad es bajo3,6,29,34. Las mutaciones en SDHC pueden estar presentes hasta en el 4% de los casos de PGL de cabeza y cuello, pero es muy raro que se asocien a PH5,8. La mutación de SDHD se asocia de forma característica a PGL parasimpáticos de cabeza y cuello, y multifocalidad; los tumores adrenales con mutaciones SDHD no suelen ser bilaterales7,15,34.
La mayor utilidad de la 18F-FDG para el diagnóstico funcional en el caso de las mutaciones SDHX y VHL y su mecanismo exacto implicado en la captación de este no se ha dilucidado. Se conoce que estos tumores tienen un aumento en la expresión de genes asociados con la angiogénesis y la hipoxia, de forma que existe una mayor activación de la glucólisis aeróbica con un aumento de fosforilación de la glucosa mediante hexocinasas que conlleva una mayor acumulación de 18F-FDG35.
Actualmente se está utilizando con más frecuencia la PET con 18F-FDG para estudiar la presencia de enfermedad metastásica, recurrencia del tumor y respuesta a la quimioterapia, aunque no está indicada para la localización inicial, debido a que su sensibilidad y especificidad es menor que otros trazadores, dependiendo de la localización y la genética (sensibilidad del 62% en SDHB negativos vs. 83% en SDHB positivos)36. En los casos de PH agresivos, que pueden ser negativos con los trazadores previamente reseñados, la 18F-FDG puede ser útil en el diagnóstico de localización de metástasis4,36.
Conclusiones
Los PH/PGL son tumores neuroendocrinos con una importante base genética y los avances en los estudios genómicos han permitido una mejor caracterización de estos tumores, que debe ser conocida en la práctica clínica diaria. Por tanto, es necesaria una priorización en el estudio genético según los datos clínicos, los antecedentes familiares, el fenotipo bioquímico y la localización del tumor.
En general, el estudio genético se recomendaría en el caso de PGL, PH adrenal bilateral, PH adrenal unilateral e historia familiar de PH/PGL, PH adrenal unilateral en menores de 30 años de edad y en todos los casos sugestivos de un síndrome familiar9,21.
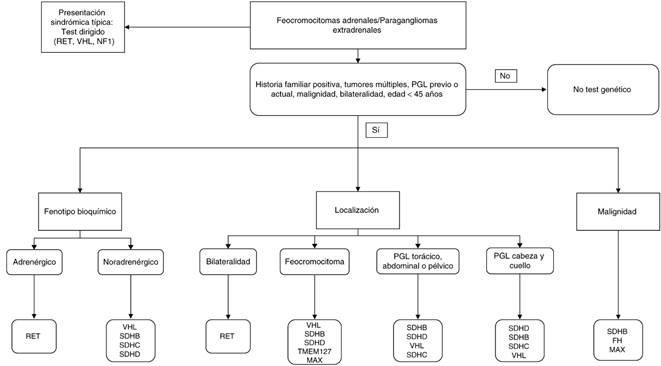
Fuente: Adaptado de: Jafri y Maher7, Galan y Kanh8.
Figura 1 Algoritmo de diagnóstico genético de PH/PGL.
Conceptos clave
El 35% de los PH y el 15% de los PGL están asociados a una mutación germinal.
No es infrecuente la historia familiar negativa, especialmente en los casos de mutaciones en SDHx, debido a la penetrancia incompleta y expresión fenotípica variable de estos genes.
La mayoría de los PH/PGL hereditarios presentan un fenotipo bioquímico específico. La metoxitiramina puede ser de utilidad en el estudio de los PGL bioquímicamente silentes o productores de DA.
El algoritmo que se muestra en la fig. 1 es una propuesta de aproximación diagnóstica genética en estos pacientes.

