










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkAnales de Medicina Interna
versão impressa ISSN 0212-7199
An. Med. Interna (Madrid) vol.22 no.7 Jul. 2005
| Enfermedad de Addison: el reto de un diagnóstico precoz E. ALEMPARTE PARDAVILA, J. L. MARTÍNEZ MELGAR, N. PIÑEIRO SANDE, Unidad de Cuidados Críticos. 1Servicios de Urgencias y 2Medicina Interna.
|
| ADDISON'S DISEASE: THE CHALLENGE OF AN EARLY DIAGNOSIS
| ||
| RESUMEN La enfermedad de Addison es un cuadro que se produce debido a la destrucción de la corteza suprarrenal, de origen principalmente autoinmune, por la presencia de anticuerpos contra la misma. Dicha destrucción tiene lugar de forma generalmente muy lenta por lo que la aparición de los síntomas es insidiosa, si añadimos el hecho de que estos síntomas suelen ser inespecíficos, es fácil entender que su diagnóstico en fases precoces sea muy difícil, con frecuencia interpretado como otra entidad clínica y sólo cuando el déficit es importante, dando lugar al desarrollo de las crisis adrenales, llegamos al diagnóstico correcto en una situación en la que el compromiso vital no es nada desdeñaable. Presentamos un caso clínico ejemplo de lo anteriormente descrito, en el que tras varios meses de clínica progresiva y varias visitas a distintos servicios médicos, se diagnostica finalmente al presentar una crisis adisoniana que hace necesario su ingreso en una Unidad de Cuidados Intensivos. PALABRAS CLAVE: Enfermedad de Addison. Insuficiencia suprarrenal. Etiología. Diagnóstico. | ABSTRACT Addison's disease is a set of symptoms which is produced due to the destruction of the suprarenal cortex, of a mainly autoimmune origin, on account of the presence of antibodies which attack it. This destruction takes place in a generally slow manner, causing symptoms to appear in an insidious way, added to the fact that these symptoms tend to be unspecific. Therefore it is easy to understand that its diagnosis at an early stage will be very difficult and is often interpreted as another disease. It is only when the deficit is important and adrenal crisis develop that we can reach the right diagnosis in a situation where the death risk is considerably high. We presented a clinical case as an example of what has been described previously. After several months of progressively deteriorating symptoms and several visits to different medical units, the patient is diagnosed as having an adrenal crisis and is immediately admitted to the intensive care unit. Key words: Addison's disease. Adrenal insufficiency. Etiology. Diagnosis. |
Alemparte Pardavila E, Martínez Melgar JL, Piñeiro Sande N, Castellano Canda P, Rodríguez García JC. Enfermedad de Adisson: el reto de un diagnóstico precoz. An Med Interna (Madrid) 2005; 22: 332-334.
Trabajo aceptado: 22 de febrero de 2005
Correspondencia: E. Alemparte Pardavila. Unidad de Cuidados Críticos. Hospital de Montecelo. Mourente, s/n. 36071 Pontevedra. e-mail: enrique.alemparte.pardavila@sergas.es
INTRODUCCIÓN
La baja incidencia de la enfermedad de Addison (EA), su lenta progresión, la inespecificidad de muchos de los síntomas de presentación y en no pocas ocasiones la ausencia de desarrollo completo de la triada típica clásica de hiperpigmentación, hipotensión e hiponatremia, hacen de esta enfermedad un auténtico reto diagnóstico. El resultado de todo ello es el deterioro de la calidad de vida del paciente incluso durante años y el riesgo añadido de verse expuesto a sufrir un evento grave como es la crisis adisoniana, de ahí la importancia de un diagnóstico lo más precoz posible que nos permita el inicio de su tratamiento, por otro lado relativamente barato y sencillo, y que nos puede evitar llegar a situaciones extremas de riesgo vital (1,2).
En el proceso diagnóstico la confirmación mediante determinaciones hormonales es fundamental, y dado que la principal etiología de la insuficiencia suprarrenal primaria es la presencia de autoanticuerpos, los test inmunológicos constituyen otro elemento de gran valor en dicho proceso (3).
CASO APORTADO
Presentamos el caso de un paciente varón de 21 años, sin alergias conocidas ni otros antecedentes, que acude a urgencias por un cuadro clínico de 6 meses de astenia, aumento de la fatiga con el ejercicio moderado y acompañado de síndrome general con disminución de peso en dichos meses de unos 10 kilos. Debido a episodios de dolor abdominal en este período acudió en dos ocasiones un Servicio de Urgencias y fue sometido a la realización de una gastroscopia informada como de mínima hernia de hiato con esofagitis grado I y reflujo biliar duodeno-gástrico. En los últimos 5 días se exacerba la clínica, presentando además vómitos, dolor abdominal difuso más intenso y deposiciones líquidas sin aumento del número de las mismas.
A la exploración física presenta una tensión arterial de 120/60 mmHg, frecuencia cardíaca de 110 pm, afebril, consciente y orientado, sin focalidad neurológica, eupneico, la auscultación cardíaca es rítmica, taquicárdica, sin soplos, el abdomen es blando y depresible, doloroso de forma difusa, sin defensa a la palpación, no se palpan masas ni megalias. Destaca la hiperpigmentación cutánea generalizada, acentuada en pezones, área genital y con manchas melánicas en paladar y bordes gingivales.
La analítica a su ingreso muestra una glucemia de 59 mg/dL, creatinina 4,32 mg/dL, urea 214 mg/dL, Na 123 mg/dL, K 8,0 mg/dL, Ca 8,2 mg/dL, una gasometría arterial con un pH 7,24, pCO2 35,6 mmHg, pO2 107 mmHg (con O2 a 4 litros/minuto), bicarbonato 14,4 mmol/L, EB - 11,3 mmol/L y SatO2 del 97,2%. El hemograma presenta una hemoglobina de 13,4 g/dl, hematocrito 39,5%, VCM 83 fL, leucocitos 12.800 (45% neutrófilos, 32% linfocitos, 12% monocitos y 10,5% eosinofilos). Coagulación sin alteraciones.
El electrocardiograma presenta una taquicardia sinusal a 110 lpm con configuración RSR' y prolongación de QT (487 ms). La radiografía de tórax es normal.
Durante su estancia en urgencias se inician medidas para la corrección de la hiperpotasemia pero el paciente progresivamente se encuentra más sudoroso, taquicárdico e hipotenso (TA 85/46 mmHg) por lo que, con la sospecha de crisis adisoniana se decide su ingreso en la UCI y se inicia la reposición de volumen con suero salino ( 4.500 cc en las primeras 24 horas), aporte de glucosa, actocortina (100 mg i.v. en bolo seguidos de 400 mg en perfusión intravenosa continua en las siguientes 24 horas); precisando inicialmente perfusión de aminas vasopresoras (dopamina a 10 mcg/kg/min), pudiendo ser retiradas en 36 horas.
Las muestras sanguíneas tomadas previas al inicio del tratamiento corticoideo para determinación de la función suprarrenal mostraban un cortisol basal 4,0 mg/dL y una ACTH en plasma 264 pg/ml.
La determinación de anticuerpos anti-cápsulas suprarrenales en suero por imnufluorescencia indirecta fue positiva con un título de 1/80.
La determinación de tirotropina, tiroxina libre, FSH y LH, asi como anticuerpos antitiroglobulina y antiperoxidasa microsomial resultaron dentro de rangos normales.
TAC abdominal: presencia de glándulas suprarrenales morfológicamente normales.
El paciente fue dado de alta de la Unidad de Cuidados Intensivos a los 3 días de su ingreso y del hospital a los 7 días con parámetros analíticos de función renal e iones dentro de la normalidad y con tratamiento con hidroaltresona 20 mg vo/8 h.
DISCUSIÓN
La enfermedad de Addison es un proceso que se desarrolla de forma lenta, y progresiva, causado por la destrucción bilateral de la corteza suprarrenal con el consiguiente déficit de la producción de hormonal que de ella depende (glucocorticoides, mineralcorticoides y andrógenos adrenales), dando lugar a un cuadro clínico inespecífico de debilidad, fatiga y pérdida de peso al que se añaden, episodios críticos de riesgo vital, las llamadas crisis adisonianas, y que representan la incapacidad de una glándula deficiente de responder a las necesidades de aumento de su producción hormonal ante situaciones de estrés (4,5).
La reserva funcional de dicha glándula hace necesaria una destrucción en torno al 90% de su tejido para que se manifieste la enfermedad, proceso que suele ser de desarrollo lento, como en el caso de las de origen autoinmune, por lo que muchos pacientes se enfrentan durante años a síntomas subclínicos, siendo su diagnostico en fases precoces difícil. De forma menos frecuente dicha destrucción se produce de forma aguda en casos de hemorragia o trombosis adrenal, sepsis o síndrome antifosfolípido (2,5,6).
Es una patología poco frecuente, generalmente infradiagnosticada, con una incidencia probablemente mayor de lo estimado, que se sitúa en torno a 0,4 a 0,8 casos por 100.000 y con una prevalencia entre 4 y 14 casos/100.000, cifras que varían según distintos estudios (3,5,7). A pesar del descenso del número de tuberculosis se mantiene esta cifra lo que hace pensar en un aumento de la incidencia de adrenalitis autoinmune: en los paises desarrollados entre el 80-90% de los casos de IAP son autoinmunes, aisladas (40%, prevalencia en varones) o formando parte del síndrome poliglandular autoinmune (60%, prevalencia en mujeres) (3).
Nuestro paciente desarrolló la enfermedad de forma gradual: el primer dato evidente fue la hiperpigmentación, el síntoma más específico de la insuficiencia adrenal primaria debido a la alta concentración de ACTH por falta de supresión del feed-back por parte del cortisol, aunque su ausencia no excluye el diagnóstico, pudiendo presentar incluso vitíligo; lo refiere progresivo en el último año y medio, con oscurecimiento difuso de la piel, pezones, con zonas más claras en la cara interna de los brazos, como puede observarse en la figura 1, y con placas azulado negruzco en borde gingibal, labios y mucosa bucal (Fig. 2).
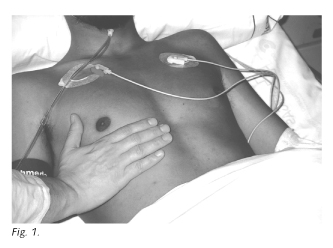
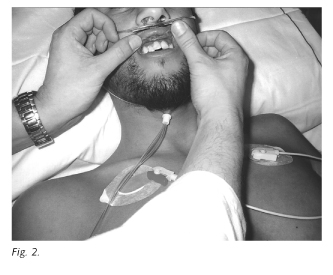
En los últimos 6 meses empieza a notar astenia, fatiga, pérdida de peso y síntomas gastrointestinales, que son la forma más frecuente de presentación de la EA, por lo que recibió atención médica en dos ocasiones, en una de ellas con gastroscopia, sin que se estableciese el diagnóstico, hecho nada raro en este tipo de patología por lo que si no se mantiene un alto índice de sospecha se nos puede pasar por alto hasta que ante un proceso intercurrente, léase infección, cirugía o trauma, el sistema suprarrenal, dañado de forma crónica y manteniendo una difícil situación de equilibrio hormonal subclínico o mínimamente evidente, se muestra incapaz de responder a las demandas de una situación de estrés y se desencadena la crisis adisoniana: instauración rápida de hipotensión, que requiere en ocasiones soporte inotrópico además de la reposición de volumen, taquicardia, fiebre, hipoglucemia y deterioro progresivo del estado mental; que precisa del aporte inmediato de las hormonas deficientes para evitar un desenlace fatal, siendo, con más frecuencia de lo deseado, en este momento cuando se establece la sospecha diagnóstica de insuficiencia suprarrenal (1,3,6,8,9).
El déficit de glucocorticoides conduce a pérdida de peso, nauseas y anorexia que se pueden acompañar de dolor muscular y articular. En ausencia de cortisol desciende el gasto cardíaco lo que provoca descenso del flujo renal y el filtrado glomerular, estimulándose la secreción de hormona antidiurética, por lo que estos pacientes son incapaces de excretar agua libre.
El déficit de mineralcorticoides, que se presenta solo en la insuficiencia adrenal primaria, produce deshidratación e hipovolemia que da lugar a tensión arterial baja, hipotensión postural y en ocasiones fallo renal prerrenal. Este déficit ocasiona además hiponatremia, hiperkaliemia y hambre de sal.
Las consecuencias hemodinámicas de todo ello son hipotensión con gasto cardíaco alto, presión capilar pulmonar normal y resistencias sistémicas bajas. El mecanismo exacto es desconocido, la patogénesis del shock en estos pacientes puede incluir la combinación de tres mecanismos: deterioro del efecto directo de los glucocorticoides sobre la musculatura vascular, que en condiciones normales produciría aumento de las resistencias sistémicas, pérdida del efecto permisivo sobre la síntesis y acción de las catecolaminas y disminución de los efectos de los glucocorticoides sobre los péptidos vasoactivos (3,4).
Ante la sospecha clínica de EA debemos obtener la confirmación bioquímica del diagnóstico, por lo que se debe tomar una muestra sanguínea para determinación de cortisol y ACTH que mostrará una elevación característica de ACTH mayor de 50 pg/ml en respuesta a niveles de cortisol prácticamente imperceptibles, menores de 5 mg/dl, aunque en las formas iniciales la cifra puede ser normal (5,6,10).
Los niveles de aldosterona están en límite bajo o por debajo del mismo con elevación de la actividad renina plasmática por encima de lo normal (3).
El diagnóstico bioquímico de la EA es fundamental previo al inicio del tratamiento de reemplazo, sin él podemos estar realizando un uso inapropiado de corticoides durante largo tiempo, por lo que el diagnóstico de la EA ante la presencia de un síndrome de fatiga crónica debe considerarse pero su diagnóstico debe realizarse de manera precisa (11).
No menos importantes, en el proceso diagnóstico de esta enfermedad, son los test inmunológicos, dado que este es el principal agente etiológico de la insuficiencia adrenal primaria. El test estándar para detectar anticuerpos frente a la corteza suprarrenal es la inmunofluorescencia indirecta, el inmunoanálisis demuestran la presencia de autoanticuerpos frente a los enzimas 17a-hidroxilasa, 21 a-hidroxilasa, con una sensibilidad en torno al 70% pero que son altamente específicos de esta enfermedad. (3,5,12).
Esta prevalencia, así como su alto valor predictivo para el desarrollo de la enfermedad ( 41% en 3 años), aconsejan la realización de screenigs en poblaciones de alto riesgo como aquellos pacientes con otras endocrinopatías autoinmunes, y el seguimiento de la función adrenal en aquellos con anticuerpos positivos (8,12).
Por otro lado, dado que la presencia de Addison autoinmune aumenta el riesgo de padecer otros trastornos autoinmunes, endocrinos o no, seria conveniente solicitar anticuerpos frente a otros órganos en el seguimiento de estos pacientes, así en alguna serie de pacientes con EA se apreció una mayor probabilidad de desarrollar enfermedad celíaca, por lo que también se sugiere que los test de diagnóstico serológico de enfermedad celíaca se incorporen en los screenings de rutina de pacientes con EA (5,14).
El estudio radiológico no tiene valor para el diagnóstico de la enfermedad, pero si de su etiología, si se sospecha infección, hemorragia, infiltración o enfermedad neoplásica se debe realizar una TAC abdominal, y debiera solicitarse tan sólo una vez confirmado el diagnóstico por determinaciones hormonales (3,5).
Como conclusión recordar, como afirma Brosnan en su artículo, que "la enfermedad de Addison sigue siendo el maestro implacable del disfraz y la inespecificidad" (1).
Bibliografía
1. Brosnan CM, Gowin NFC. Addison's disease. BMJ 1996; 312: 1085-1087. [ Links ]
2. Baker S, Kenward D, White KG. Addison's disease: after 40 years much remains the same. BMJ 2001; 322: 494. [ Links ]
3. Arit W, Allolio B. Adrenal insufficiency. Lancet 2003; 361: 1881-1893. [ Links ]
4. Werbel SS, Patrick K. Acute adrenal insufficiency. Endocrinol Metab Clinics North Am 1993; 22: 303-328. [ Links ]
5. Candel González FJ, Matesanz David M, Candel Monserrate I. Insuficiencia corticosuprarrenal primaria. Enfermedad de Addison. An Med Interna (Madrid) 2001; 18: 492-498. [ Links ]
6. Oelkers W. Adrenal insufficieny. Current-concepts. N Engl J Med 1996; 335: 1206-1212. [ Links ]
7. Laurenti S, Vecchi L, Santeusanio F, Falorni A. Is the prevalence of Addison's disease underestimated? J Clin Endocrinol Metab 1999; 84: 1762. [ Links ]
8. Ten S, New M, Maclaren N. Clinical review 130: Addison's disease 2001. J Clin Endocrinol Metab 2001; 86: 2909-2922. [ Links ]
9. Keljo D, Squires R. Just in time. N Engl J Med 1996; 334: 46-48. [ Links ]
10. Leman P. Hydrocortisone should be started immediately adrenal insufficiency is considered. BMJ 1996; 313: 427. [ Links ]
11. Robinson S, Grossman A. Addison's disease should be diagnosed biochemically. BMJ 2001; 323: 51. [ Links ]
12. Baker JR. Autoimmune endocrine disease. JAMA 1997; 278: 1931-1937 [ Links ]
13. Martín Martorell P, Roep BO, Smit JW, Martorell PM. Autoimmunity in Addison's disease. Neth J Med 2002; 60: 269-275. [ Links ]
14. O'leary C, Walsh CH, Wieneke P, O'regan P, Buckley B, O'halloran DJ, et al. Coeliac disease and autoimmune Addison's disease: a clinical pitfall. Q J Med 2002; 95: 79-82. [ Links ]

