










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista de Diagnóstico Biológico
versión impresa ISSN 0034-7973
Rev Diagn Biol vol.50 no.3 jul./sep. 2001
REVISIÓN
La Citometría de Flujo en el Análisis de las Plaquetas.
(I) Aspectos Estructurales y Funcionales de las Plaquetas
M.C. Monteiro*, J.E. O´Connor**, M. Martínez***
* Departamento de Bioquímica, Instituto Politécnico de Saúde-Norte, Gândra
** Centro de Citometría, Departamento de Bioquímica y Biología Molecular,
Facultad de Medicina y Odontología, Universidad de Valencia.
*** Departamento de Biopatología Clínica, Hospital Universitario La Fe, Valencia.
Palabras clave: Plaquetas; activación; agregación plaquetaria;glicoproteínas; calcio; citoesqueleto.
Keywords: Platelets; activation; platelet aggregation; glycoproteins; calcium; cytoskeleton.
Recibido: 10-I-01
Aceptado: 22-IV-01
Correspondencia: María-do-Céu Monteiro.
Departamento de Bioquímica, Instituto Politécnico da Saúde-Norte,
Rua Central de Gândra 1317.
4585-116 Gândra PRD, Portugal
SUBVENCIONES ECONÓMICAS:
Ayuda PRODEP, Ministério de Educaçâo, Portugal, para la realización de los estudios de doctorado de María do Céu Monteiro.
Convenio Universidad de Valencia-Izasa, S.A. para la Creación y Mantenimiento de un Centro de Citometría en la Universidad de Valencia.
Primer Premio Laboratórios LabMed, Portugal.
1. Características generales de las plaquetas
Las plaquetas son pequeñas células anucleadas, procedentes de los megacariocitos y en condiciones fisiológicas normales tienen la forma de disco biconvexo, con un diámetro aproximado de 3 µm. De la masa plaquetaria total, cerca de 2/3 circulan en la sangre, encontrándose las restantes retenidas en el bazo.
Aunque inicialmente han sido descritas como pequeñas partículas inertes, hoy se sabe que son células con una extraordinaria complejidad estructural y bioquímica que se expresa en la gran variedad de funciones que desempeñan. Han sido incluso utilizadas como modelos para el funcionamiento de neuronas, debido a su capacidad de liberar y captar serotonina y otros neurotransmisores
Las plaquetas desempeñan un papel fundamental en la hemostasia, interviniendo en el mecanismo fisiológico que protege el organismo de la pérdida exagerada de sangre, como consecuencia de una lesión de los vasos en que haya ruptura o alteración del endotelio vascular, sobre todo en el territorio arterial.
En condiciones normales, las plaquetas circulantes no se adhieren a la superficie endotelial, ni entre ellas, debido al equilibrio existente entre los mecanismos pro- y anti-trombóticos. Cuando hay una lesión, se adhieren a estructuras subendoteliales expuestas, son activadas y se agregan unas a otras, constituyendo, así, una parte esencial del tampón hemostático primario.
Diversas sustancias pueden activar las plaquetas originando una o más de las respuestas características, íntimamente asociadas en la hemostasia: adhesión, cambio de forma, agregación y secreción.
Además de formar un agregado en el lugar de la lesión, las plaquetas, cuando son estimuladas, sufren alteraciones en la membrana que originan la exposición de una superficie pro-coagulante, que promueve la activación de algunos factores de la coagulación, incluyendo la protrombina y el factor X, y liberan inhibidores de la fibrinólisis, los cuales retrasan la destrucción de las mallas de fibrina por las enzimas fibrinolíticas1,2,3.
Los hallazgos de Bizzozero4 permitieron el reconocimiento del papel de la plaqueta en la hemostasia y constituyeron la base inicial de los estudios que han dado lugar a los conocimientos hoy existentes.
Además de su actividad fisiológica principal de participación en las reacciones hemostáticas, las plaquetas intervienen en otro tipo de procesos fisiológicos o patológicos, como la inflamación, la cicatrización de heridas, la trombosis, la aterosclerosis o metástasis tumorales1.
Lo que es, en condiciones normales, una respuesta fisiológica puede, en condiciones patológicas, llevar a la formación de trombos en los cuales las plaquetas, junto a la formación de fibrina, podrán ocluir un vaso. Ejemplo de este tipo de patologias son las enfermedades tromboembólicas, principal causa de morbilidad y mortalidad de los países desarrollados. Así, el conocimiento de la fisiología y patología plaquetaria constituye un área fundamental de la investigación de la hemostasia.
1.1 Estructura de las plaquetas
Los principales orgánulos de la plaqueta son la membrana plasmática, el citoesqueleto, el sistema canalicular abierto, el sistema tubular denso y los gránulos. Sin embargo, están presentes otros, incluyendo mitocondrias y gránulos de glucógeno.
A continuación, se describen los principales aspectos de estas estructuras relacionados con su función.
1.1.1 Membrana plasmática
La membrana plasmática de la plaqueta, como ocurre con otras células, media en las interacciones con el medio externo, ocupando, por eso, un papel central en su fisiología. Es una típica unidad de membrana con un contenido particular en determinados fosfolípidos, glicolípidos y glicoproteínas.
Los fosfolípidos son particularmente ricos en ácido araquidónico, el ácido graso precursor de la síntesis de eicosanoides, sustancias implicadas en la transmisión de las señales recibidas en la membrana. Los fosfolípidos se distribuyen asimétricamente en la bicapa y los que poseen carga negativa, tales como el fosfatidilinositol, fosfatidilserina y fosfatidiletanolamina se localizan preferentemente en la capa interna5. Esta distribución tiene, también, un significado funcional, ya que sirven como sustratos para enzimas intracelulares (fosfolipasas) durante la activación plaquetaria. Algunos fosfolípidos y sus derivados tienen propiedades agregantes, sobre todo el factor activador de plaquetas (PAF), o actividad procoagulante, como es el caso del factor plaquetario 3 (PF3), y se tornan accesibles cuando la plaqueta es activada, debido a una reorganización de los componentes de la membrana6. La membrana plaquetaria contiene un gran número de glicoproteínas con una o más cadenas ramificadas de polisacáridos, que forman una cubierta exterior o "glicocálix", que confiere una carga negativa a la superficie de la plaqueta7.
La membrana plasmática tiene una composición en glicoproteínas variable (Fig.1). Durante el proceso de secreción, se produce la fusión de las membranas de los gránulos intraplaquetarios con la membrana plasmática a través del sistema canalicular abierto, lo que permite la exposición de antígenos internos en la superficie de la plaqueta. Glicoproteínas como la GMP140, que existe sólo en las membranas de los gránulos α, y la GP53 de la membrana de los lisosomas de las plaquetas en reposo, pasan a expresarse en la superficie después de la activación y secreción de las plaquetas. Así, la composición de la membrana plasmática depende del estado de activación de la plaqueta8,9.
Las glicoproteínas clásicas han sido subclasificadas en distintas familias: integrinas, glicoproteínas ricas en leucina y selectinas. Las glicoproteínas de la membrana plaquetaria actúan como receptores, mediando, entre otras, en tres importantes funciones: en la adhesión de las plaquetas a componentes de la matriz extracelular de la pared vascular, en la agregación plaquetaria y en la interacción de las plaquetas con otras células10. Diversos receptores han sido ya clonados, secuenciados e identificados11,12,13, y sus características bioquímicas y funcionales se resumen en la Tabla 1.
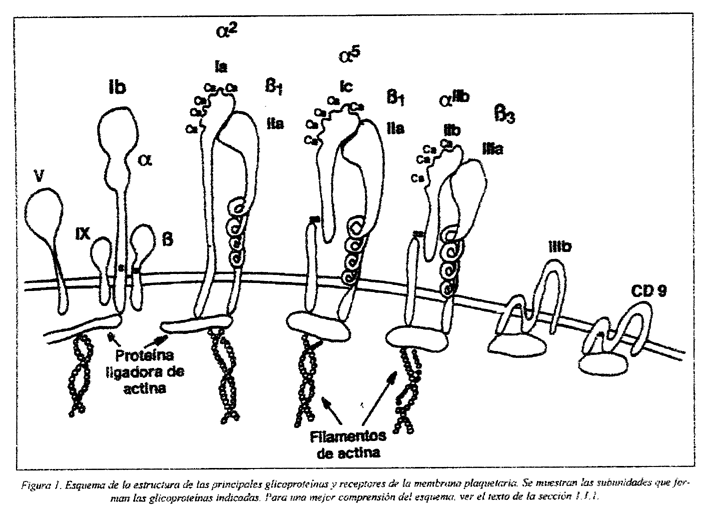
1.1.1.1 Receptores de la familia de las integrinas
Las integrinas son una familia de glicoproteínas de la membrana implicadas en interacciones célula-matriz o célula-célula.
Son heterodímeros constituidos por dos subunidades, α y ß, y para la mayoría de estos receptores el lugar de reconocimiento en el ligando es el tripéptido Arg-Gly-Asp (RGD). Este grupo incluye cinco receptores de la membrana plaquetaria: GP IIb/IIIa, GP Ia/IIa, GP Ic/IIa, GP Ic'/IIa y el receptor de la vitronectina14,15,16.
a) GPIIb/IIIa:
El complejo GPIIb/IIIa es la integrina más abundante y se encuentra en forma de heterodímeros αIIßb3, dependientes de calcio. Su función principal es la de receptor para el fibrinógeno, mediando la agregación plaquetaria.
Las plaquetas normales contienen alrededor de 50.000 complejos GPII/IIIa en la membrana plasmática; no obstante, otros de estos complejos están presentes en las membranas del sistema canalicular abierto y de los gránulos a, y pueden expresarse en la superficie, después de la activación de las plaquetas12,17.
El complejo GPIIb/IIIa actúa como receptor para otras proteínas adhesivas, además del fibrinógeno, tales como la fibronectina, el factor de von Willebrand (vWF) y la vitronectina. A través de uniones con estas proteínas adhesivas, interviene en el proceso de adhesión al subendotelio. Estos complejos solamente adquieren capacidad para interactuar con las proteínas adhesivas, después de la activación de la plaqueta. Tras la activación hay un incremento del número de complejos expuestos en la superficie, así como un incremento de su actividad resultante de alteraciones conformacionales que conducen a la exposición de los lugares de unión. El reconocimiento de los ligandos por el heterodímero implica la secuencia RGD y requiere Ca2+ y Mg2+17,18,19.
La unión al fibrinógeno induce otras alteraciones conformacionales en el complejo, las cuales son responsables de la transmisión de señales al interior de la plaqueta. La participación del complejo en la transducción de la señal después de la activación celular, incluye la regulación de la fosforilación de tirosinas, intercambio de Na+/H+, aglomeración de proteínas del citoesqueleto y entrada de calcio a través de la membrana. Por otra parte, el complejo GPIIb/IIIa interviene en la retracción del coágulo, uniendo la red de fibrina extracelular al aparato contráctil intracelular20,21,22,23.
Por todo lo expuesto, podemos concluir que la presencia de GPII/IIIa es imprescindible para que haya agregación plaquetaria y hemostasia normal. Este hecho se evidencia en la enfermedad hemorrágica congénita designada por trombastenia de Glanzmann24,25,26 determinada por la falta de este complejo funcional.
b) GPIa/IIa:
GPIa/IIa es el receptor para el colágeno, corresponde al heterodímero α2ß1 y está presente en la plaqueta en un número aproximado de 2.000 moléculas por célula. Este receptor funciona en el proceso de adhesión a la matriz extracelular, y también está implicado en la agregación de las plaquetas inducida por el colágeno. La unión al colágeno depende de Mg2+ y Mn2+ 27,28.
C) GPIc/IIa, GP Ic'/IIa y el receptor de la vitronectina:
GPIc/IIa, G Ic'/IIa y el receptor de la vitronectina, corresponden a las integrinas minoritarias de la membrana plaquetaria α5ß1, α6ß y αVß, y son, respectivamente, los receptores para la fibronectina, laminina y vitronectina27,28,29.
1.1.1.2 Glicoproteínas ricas en leucina (LRG)
A la familia de las glicoproteínas ricas en leucina pertenecen proteínas con funciones muy diversas, que tienen en común la presencia de un número variado de copias de un segmento, con una secuencia de 24 residuos de aminoácidos rica en leucina (LRG). En este grupo está incluido el complejo GP Ib/IX/V de la membrana plaquetaria27,30,31.
a) GPIb/IX/V:
El complejo glicoproteico GPIb/IX/V es un receptor específico implicado en la adhesión y agregación plaquetaria. Es el principal responsable de la adhesión de las plaquetas al subendotelio en zonas vasculares donde prevalecen condiciones de flujo caracterizadas por una elevada tensión de cizallamiento. Éste complejo interactúa con las estructuras subendoteliales, principalmente con el colágeno, a través de un ligando adhesivo, el factor de von Willebrand (vWF). El vWF es una proteína multimérica constituyente de la matriz subendotelial, que está presente en los gránulos a de la plaqueta, desde donde es secretado durante la activación, y circula en la sangre formando un complejo con el factor VIII de la coagulación (FVIII:c)32,33.
El receptor GPIb/IX/V también funciona como lugar de unión de alta afinidad para la trombina, participando en la propagación de la respuesta a este importante agonista en la activación y agregación plaquetaria31, 34.
Se estima que el número de moléculas de este complejo se aproxima a 25.000 / plaqueta, siendo la segunda glicoproteína más abundante de la membrana citoplasmática. La GPIb es la principal sialoglicoproteína de la membrana de las plaquetas y, como tal, es responsable en gran medida de la carga negativa de las plaquetas y de su comportamiento electroforético7,34.
Aproximadamente el 70% de la GPIb total de la membrana está asociada a la actina y esta asociación al citoesqueleto puede estar relacionada, tanto con el mantenimiento de la forma de la plaqueta, como con la función de unión del vWF. En las plaquetas en reposo, el complejo está unido al citoesqueleto a través de la proteína ligadora de actina (ABP) y su exposición en la superficie disminuye después de la activación35,36,37.
La fosforilación de receptores de la membrana puede constituir un importante mecanismo de transducción de la señal después de la unión del respectivo ligando. Sin embargo, la fosforilación de esta proteína no parece estar implicada en la transmisión de la señal ni en la activación plaquetaria. Al contrario, existen evidencias de que la fosforilación de la serina 166 en el dominio citoplasmático de la GP Ibb puede ser, en parte, responsable del mantenimiento de la plaqueta en estado de reposo (34, 35, 38).
El complejo GPIb/IX/V es esencial para la normal adhesión y activación plaquetaria, lo que se evidencia en los pacientes deficientes en este complejo. Tales alteraciones resultan de anomalías congénitas, conocidas como síndrome de Bernard-Soulier, y conducen a una tendencia hemorrágica, la cual puede ser extremamente severa39,40.
1.1.1.3 Otros receptores:
a) GPIV:
La GPIV es una glicoproteína mayoritaria de la membrana plaquetaria y funciona como receptor para la trombospondina, y para las fibrillas de colágeno del tipo I12. Existen alrededor de 23.000 moléculas de GP IV por plaqueta, encontrándose la mayor parte de éstas en la membrana citoplasmática de la plaqueta en reposo. Recientemente se demostró la presencia de esta glicoproteína en la membrana de los gránulos a, y el aumento de su expresión en la superficie después de la estimulación de las plaquetas con trombina41,42.
1.1.2 Sistema canalicular abierto
El sistema canalicular abierto (SCA) se presenta como una red de vesículas y canales, interconectados, que se ramifican a través de todo el citoplasma y comunican con la superficie. Tienen una localización preferencial bajo la membrana celular y están aparentemente desprovistos de contenido.
El SCA consiste en invaginaciones de la membrana citoplasmática de la cual deriva su estructura, estando la cara interna de estas vesículas revestida por un depósito floculoso idéntico al glicocálix. Este sistema constituye una vía de acceso de sustancias plasmáticas a lugares más internos de la célula, y durante la activación plaquetaria constituye una reserva de membrana plasmática que permite el cambio de forma y la emisión de pseudópodos. Funciona además como conductor hacia el exterior de sustancias liberadas a partir de los gránulos5,6.
1.1.3 Sistema Tubular Denso
Al microscopio electrónico (ME) el sistema tubular denso (STD) se presenta como un conjunto de tubos apretados y cortos, que se distinguen del SCA por su opacidad, similar al del citoplasma que los circunda. Estos tubos forman una red continua por todo el citoplasma, siendo más apretada en la periferia que en el centro de la célula. El STD puede estar en asociación íntima, tanto con los microtúbulos, como con el sistema canalicular abierto. Sus membranas derivan del retículo endoplasmático de los megacariocitos6.
El STD constituye el principal lugar de almacenamiento del calcio intraplaquetario, siendo por eso comparable al retículo sarcoplásmico de las células del músculo estriado, y el SCA puede considerase equivalente a los túbulos transversos de las referidas fibras musculares.
El STD acumula Ca2+ debido a la presencia de un transportador Ca2+ ATPasa del tipo SERCA, en sus membranas, que regula la concentración de Ca2+ citoplasmático libre43. Similarmente a lo que ocurre en otras células, innumerables fenómenos de la fisiología plaquetaria dependen de la concentración citoplasmática de este ion (Tabla 2)44,45. La actividad de este transportador es esencial, por ejemplo, para mantener los microtúbulos en la forma polimerizada y, consecuentemente, la forma discoide de la plaqueta, que requiere una baja concentración citoplasmática de Ca2+.
Las membranas del STD son particularmente ricas en los fosfolípidos, que funcionan como sustratos para la síntesis de endoperóxidos de prostaglandinas y tromboxano A2, y en él también se alojan las enzimas respectivas6.
1.1.4 Citoesqueleto
El citoesqueleto consiste en una red de estructuras filamentosas que mantienen la estructura de la plaqueta, estando la reorganización de estas estructuras implicada en las respuestas de la plaqueta a la activación. Contiene las proteínas contráctiles actina y miosina, las proteínas implicadas en la formación de los microtúbulos, principalmente la tubulina, y otras asociadas a éstas5,35.
La actina se encuentra, en las plaquetas no activadas, tanto en la forma polimerizada, actina-F (40-50%), como en la forma monomérica, actina-G. Los monómeros poseen un peso molecular de 44.000 y pueden ser del tipo α o γ. La polimerización es impedida, esencialmente, por la profilina, que forma un complejo con la G-actina, y por la gelsolina, que protege la extremidad de los polímeros ya formados35.
Cuando las plaquetas son activadas, una cantidad adicional de actina polimeriza y se asocia a otras proteínas, tales como, la tropomiosina, la a-actinina y la ABP, lo que determina la organización de estos filamentos en una red tridimensional periférica y la formación de haces, originando los pseudópodos. Estas transformaciones de la actina son responsables del cambio de forma de las plaquetas, el cual caracteriza su activación en el aspecto morfológico. La contracción de los filamentos periféricos hace que los gránulos dispersos en el citoplasma ocupen una posición central, y simultáneamente liberen sus contenidos vía SCA.
La ABP une los filamentos de actina a diversas proteínas de la membrana plasmática: complejo GPIb/IX/V, GPIa, GPIIa y una proteína recientemente identificada de un peso molecular de 250.000. Después de la activación plaquetaria parte de estas glicoproteínas, especialmente la GPIb, deja de estar asociada al citoesqueleto debido a la hidrólisis de la ABP por una proteasa dependiente de Ca2+. A la inversa, el complejo GPIIb/IIIa se encuentra asociado a los filamentos de actina de los pseudópodos después de la agregación, y no en las plaquetas en reposo. La unión del fibrinógeno al complejo GPIIb/IIIa induce en éste alteraciones conformacionales que lo llevan a unirse al citoesqueleto vía talina y vinculina35,46.
La fuerza contráctil es generada cuando la miosina es fosforilada e interactúa con los filamentos de actina. La miosina plaquetaria, tal como la miosina del músculo esquelético, está constituida por 6 cadenas polipeptídicas, similares dos a dos. La actividad ATPásica de la miosina se desencadena por la fosforilación de las cadenas ligeras, permitiendo la interacción con la actina. El funcionamiento del sistema actina-miosina plaquetario es, en la mayor parte de sus aspectos, similar al observado en el músculo esquelético.
En las plaquetas en reposo, los microtúbulos son los únicos componentes del citoesqueleto visibles al microscopio electrónico. Son estructuras tubulares constituidas por 13 subfilamentos de tubulina y cada subfilamento es una sucesión alternada de tubulinas α y β, proteínas globulares de peso molecular 55.000. Parecen estar dispuestos en haces de 8 a 24 anillos de microtúbulos pero, en realidad, se trata de uno sólo enrollado en espiral, muy cerca de la membrana celular, siguiendo la circunferencia mayor de la plaqueta. El haz de microtúbulos, responsable del mantenimiento de la forma discoide de las plaquetas en reposo, se deforma durante el proceso de activación, se fragmenta transitoriamente y se reensambla en una posición más central, circundando los gránulos plaquetarios5,35.
1.1.5 Gránulos
La plaqueta contiene, distribuidos por su citoplasma, un gran número de gránulos, delimitados por una membrana unitaria, que se pueden distinguir por sus contenidos específicos. Normalmente son reconocidos cuatro tipos: los gránulos a, los gránulos densos, los lisosomas y los microperoxisomas. En la plaqueta activada, estos gránulos son centralizados, a lo que sigue la secreción de sus contenidos.
Los gránulos α constituyen la gran mayoría de estas estructuras (cerca del 85%). Poseen en su interior una gran diversidad de proteínas, algunas de las cuales son específicas de la plaqueta, otras, son homólogas a proteínas plasmáticas y tisulares (Tabla 3). En la fase de secreción estos factores son liberados en las proximidades de la plaqueta y participan, sobre todo, en la hemostasia, produciendo un efecto pro-coagulante, estimulando la adhesión y la agregación, y favoreciendo procesos de reparación de los vasos lesionados.
Las proteínas de la matriz de los gránulos a tienen dos orígenes distintos: algunas son sintetizadas por los megacariocitos y empaquetadas en los gránulos por el complejo de Golgi, como por ejemplo el vWF, factor V y la trombospondina. Otras proteínas, tales como el fibrinógeno, que también se encuentra en circulación, son endocitadas a partir del medio externo e incorporadas al interior de los gránulos α. Este fenómeno puede ser mediado por receptores de membrana, como es el caso de la incorporación del fibrinógeno en que está implicado el complejo GP IIb/IIIa47.
Hay tres proteínas que se consideran exclusivas de las plaquetas: el factor plaquetario 4 (PF4), la ß-tromboglobulina (ß-TG) y el factor de crecimiento derivado de la plaqueta (PDGF).
El PF4 es una glicoproteína monomérica, muy parecida estructuralmente a la ß-TG con la que presenta diversas secuencias de aminoácidos homólogas. Ambas proteínas pueden combinarse con la heparina neutralizando su efecto anticoagulante, pero la ß-TG tiene una afinidad más baja para este polisacárido48.
El PDGF es una proteína catiónica constituida por dos cadenas polipeptídicas unidas por puentes disulfuro. Posee actividad mitogénica para diferentes líneas celulares, incluyendo células del músculo liso, endoteliales y de la glía. Este factor está implicado en la reparación de la pared del vaso lesionado, produciéndose su liberación después de la adhesión de la plaqueta al subendotelio5.
La membrana de los gránulos α también contiene proteínas que se expresan en la superficie de la célula después de la activación. Entre éstos se encuentran el complejo GPIIb-IIIa, la GMP140 y la osteonectina49.
Los gránulos densos presentan una gran opacidad al microscopio electrónico en la zona central, atribuida a la presencia de Ca2+. Además de Ca2+, contienen serotonina, ADP, ATP y pirofosfato y pueden captar dopamina a partir del exterior.
Los lisosomas se caracterizan por su contenido rico en enzimas hidrolíticas, principalmente hidrolasas ácidas. La exteriorización del contenido de estos gránulos puede ser observada durante la activación plaquetaria con agonistas fuertes, por ejemplo a través de la exposición en la superficie celular de glicoproteínas, solamente presentes en la membrana de los lisosomas de la plaqueta en reposo.
1.1.6 Otros orgánulos plaquetarios
La plaqueta presenta, además de estos gránulos implicados directamente en la función hemostática, otros orgánulos esenciales a su metabolismo, incluyendo mitocondrias, gránulos de glucógeno e inclusiones lipídicas. La plaqueta está adaptada para disponer rápidamente grandes cantidades de energía, principalmente durante los procesos de agregación, secreción y retracción del coágulo. Presenta, por eso, la vía glicolítica muy activa, así como la biosíntesis y degradación del glucógeno. Cuando está en reposo, la plaqueta puede producir ATP por fosforilación oxidativa al nivel de la membrana mitocondrial. La matriz de la mitocondria, además de contener las enzimas del Ciclo de Krebs posee las de la ß-oxidación de los ácidos grasos50.
1.2. Origen y producción de las plaquetas
Las plaquetas son pequeña células anucleadas, con origen en los megacariocitos. En condiciones fisiológicas normales, no activadas, tienen la forma de disco biconvexo, con un diámetro aproximado de 3 µm. Se encuentran en circulación en una concentración entre 150.000 y 450.000 plaquetas/µl y tienen una vida media de 7 a 10 días, siendo renovadas a una velocidad de 35.000/µl/día6.
Los megacariocitos originan las plaquetas por desprendimiento de fragmentos de su citoplasma. Estas grandes células de la médula ósea, son descendientes de una célula pluripotente diploide, la stem cell hemopoyética, que puede formar precursores eritroides, granulocíticos y megacariocíticos. Su diferenciación y proliferación hasta la línea megacariocítica es un proceso regulado por numerosas citokinas y factores de crecimiento hematopoyéticos.
Los progenitores megacariocíticos son las BFU-Meg (unidades formadoras de burst megacariocíticos) y las CFU-MK (unidades formadoras de colonias megacariocíticas). En la serie megacariocítica, después de un período en que las células progenitoras unipotentes van a sufrir varias mitosis, se inicia un proceso de endomitosis sucesivas. Los precursores, megacarioblastos, que eran diploides (2N) pasan a presentar una ploidia variable. Este estadio se caracteriza por un crecimiento del volumen celular, un desarrollo del sistema de membranas y síntesis de gránulos específicos. Al final de esta fase, encontramos células con ploidias de 4N a 64N, siendo en condiciones fisiológicas la ploidía modal de la especie humana entre 16N y 32N51. El grado de ploidía alcanzado condiciona el tamaño celular y éste el número de plaquetas que cada célula es capaz de producir.
Además del nucleo poliploide, la característica ultraestructural más importante de los megacariocitos es el desarrollo del sistema de membranas de demarcación (SMD).
Dado que las plaquetas maduras son anucleadas, la síntesis de la mayoría de las proteínas plaquetarias ocurre durante la maduración del megacariocito. Adicionalmente, éste tiene capacidad de captar proteínas plasmáticas circulantes y almacenarlas en la matriz de los gránulos en desarrollo52. Los megacarioblastos y promegacariocitos, con intensa actividad biosintética, muestran un citoplasma integrado principalmente por ribosomas y trayectos de reticulo endoplasmático rugoso junto a mitocondrias, vesículas de Golgi y unas pocas estructuras dispersas típicas del megacariocito maduro, como las membranas de demarcación y los gránulos α, con tendencia a aumentar en las fases más tardías.
Los megacariocitos han mostrado alguna capacidad de activación al ser estimulados por agonistas plaquetarios. Cuando son activados por la trombina, el ADP, la epinefrina o el ionóforo de Ca2+ A23187, según el estímulo y su concentración, los megacariocitos cambian de forma con aumento del área celular y traslado del núcleo a un polo de la célula, contracción celular y reacción de liberación de los gránulos densos o, en determinados casos, disrupción de los megacariocitos en fragmentos tras la contracción53.
Los mecanismos implicados en la fase final de generación y liberación de las plaquetas no están aún bien establecidos54. La mayoría de los investigadores consideran que al final de la maduración, los megacariocitos localizados en zonas subendoteliales de los capilares medulares, emiten proyecciones citoplasmáticas (pseudópodos) hacia el interior de éstos. Las plaquetas son liberadas directamente en la corriente sanguínea por la ruptura de áreas de constricción que aparecen a lo largo de los pseudópodos55,56. Otra hipótesis para el mecanismo de liberación de las plaquetas propone que las membranas de demarcación delimitan territorios plaquetarios en el citoplasma y que las plaquetas se liberan después de la fragmentación de la membrana. Algunos megacariocitos pueden alcanzar la circulación y permanecer en los vasos pulmonares donde pueden formar plaquetas57.
El número de plaquetas de un individuo tiende a mantenerse constante. Cualquier alteración del número de plaquetas como consecuencia de la inducción de trombocitopenia o de una transfusión de plaquetas, es seguida de alteraciones en la producción de plaquetas, que tiende a restablecer el recuento normal. El principal factor humoral responsable de la regulación de la producción de plaquetas, es la trombopoyetina (TPO)55,58,59,60.
Además de la TPO, existen otros reguladores hematopoyéticos con acción proliferativa en los megacariocitos incluyendo, las interleukinas IL-3, IL-6, IL-11, el factor inhibidor de la leucemia y GM-CSF (Granulocyte-Macrophage Colony Stimulating Factor). Estos reguladores, a pesar de tener la capacidad de incrementar los niveles de plaquetas, son de acción más lenta y afectan a una gran variedad de células58. Por otra parte, la megacariocitopoyesis es inhibida por reguladores negativos incluyendo el PF4 y el TGFß1(Transforming Growth Factor ß1)61.
En la población total de plaquetas pueden separarse sub-poblaciones con diferentes tamaños. Las plaquetas mayores, los megatrombocitos, difieren de las otras en diversas características, principalmente en el contenido en gránulos, en las propiedades de adhesión y agregación. Alteraciones en el número relativo de megatrombocitos, pueden explicar alteraciones de la bioquímica de la función plaquetaria en circunstancias fisiológicas, o de diversas disfunciones asociadas a un recambio plaquetario anormal.
Las plaquetas con diferentes tamaños pueden tener origen en diferentes poblaciones de megacariocitos, lo que se designa por heterogeneidad intrínseca. Disfunciones en las cuales el contenido de DNA en los megacariocitos está aumentado, están asociadas a un incremento del tamaño plaquetario, mientras que disfunciones con una disminución del DNA magacariocítico llevan a la producción de plaquetas menores50. Por otro lado, las plaquetas disminuyen de tamaño y sufren alteraciones de su función durante el envejecimiento, lo que determina la heterogeneidad adquirida62,50. Recientemente se estudió la relación entre el contenido de RNA plaquetario y el tamaño, y se verificó que hay una relación lineal63.
2. Activación plaquetaria
2.1 Mecanismo de activación: vías de transducción de la señal
Los agonistas fisiológicos de los que se conoce su implicación en la activación plaquetaria incluyen macromoléculas de la matriz subendotelial (colágeno, vWF), hormonas circulantes (adrenalina y vasopresina), sustancias generadas en la lesión (trombina) o por plaquetas activadas (tromboxano A2, ADP, serotonina), y sustancias producidas por otras células (factores de activación plaquetaria). Para contrarrestar estos estímulos positivos, las células endoteliales producen sustancias inhibidoras de la activación de las plaquetas, tales como la PGI2, y el oxido nítrico.
La activación se inicia con la unión del agonista a receptores de la membrana plasmática plaquetaria. Esta interacción resulta en la modulación de la actividad de canales iónicos, enzimas asociadas a la membrana y citoplasmáticas, que generan segundos mensajeros, transmisores de la señal de activación a sistemas efectores responsables de las manifestaciones externas de activación de la plaqueta.
La adhesión inicial de las plaquetas a un vaso lesionado es un proceso pasivo, pero esta unión resulta en la activación celular y favorece esa adhesión. La agregación plaquetaria, al contrario de la adhesión, es totalmente dependiente de las alteraciones inducidas por la activación celular, particularmente, de la exposición de lugares de unión para el fibrinógeno. La secreción de los gránulos, a su vez, requiere la activación de la maquinaria contráctil y la fusión de las membranas de los gránulos, con las del SCA y con la membrana plasmática.
Similarmente a otras células, en la plaqueta las proteínas ligadoras de nucleótidos de guanina (proteínas G) funcionan como elementos transductores de señales extracelulares, desde los receptores hasta enzimas celulares y canales iónicos22,64,65,66.
Las proteínas G son heterotrímeros constituidos por las subunidades Gα, Gß y Gγ. y la subunidad Gα contiene el lugar de unión de los nucleótidos de guanina (GDP y GTP). En su estado inactivo, la proteína G se encuentra unida a GDP. Cuando el receptor es activado, el GDP es sustituido por GTP y la subunidad Gα-GTP se disocia de las subunidades Gßg. La activación e inhibición de enzimas diana y canales implica alteraciones conformacionales inducidas, en parte, por GTP, y que están íntimamente relacionadas con la disociación de Gα de Gßγ y con alteraciones de la estructura secundaria y terciaria de la Gα. La actividad GTPásica intrínseca de esta subunidad lleva a la hidrólisis lenta del GTP y consecuente formación de GDP. La Gα-GDP vuelve a unirse a las subunidades Gßg, terminando el ciclo desencadenado por el agonista67. Las proteínas G son clasificadas, según las formas de Gα, en cuatro familias principales, representadas por las subunidades Gsα, Giα, Gqα y G12α, habiendo ya sido identificadas en la plaqueta nueve proteínas G68,69.
Los receptores para muchos agonistas de las plaquetas interactúan con las proteínas G para activar fosfolipasas, tales como la fosfolipasa C (PLC) y fosfolipasa A2 (PLA2), tirosina-kinasas y también suprimiendo la formación de AMPc por inibición de la adenilciclasa. Como el AMPc tiene un papel inhibidor en la plaqueta, ambos efectos favorecen la activación plaquetaria66,70,71.
La hidrólisis enzimática de fosfolípidos específicos de la membrana se lleva a cabo por dos vías, una mediada por PLC y otra por PLA2, las cuales determinan la formación de segundos mensajeros activadores de vías bioquímicas intracelulares comunes.
La fosfolipasa C inicia el metabolismo de los fosfoinositidos y los productos de su reacción incluyen inositol-1,4,5-trifosfato (IP3) y diacilglicerol (DG)72. El IP3 promueve la liberación de Ca2+ del STD, contribuyendo al incremento de la concentración intracelular de este ion73,74,75,76. Un aumento en el Ca2+ citoplasmático, tiene un papel central en la activación de la plaqueta estimulando la respuesta de sistemas dependientes, tanto de Ca2+ libre, como del complejo Ca2+ calmodulina35,44,77,78,79,80. El DG activa la proteína-kinasa C, llevando a la fosforilación de proteínas, secreción de los gránulos y expresión del receptor para el fibrinógeno81,82,83.
La fosfolipasa A2 media en la formación de endoperóxidos de prostaglandinas y de tromboxano A2 (TxA2), liberando gran cantidad de ácido araquidónico (AA) a partir de fosfolípidos de la membrana, durante la activación plaquetaria84. Una vez liberado, el AA es convertido en endoperóxidos de prostaglandinas (PGs) y TxA2 por la vía de la cicloxigenasa. Los endoperóxidos de PGs y TxA2 amplifican la activación de la plaqueta tanto por la interacción con receptores específicos en la membrana de la plaqueta que están unidos a la PLC, como induciendo la secreción de los gránulos α y de los gránulos densos66,85.
Las investigaciones más recientes de la activación plaquetaria han evidenciado que la señalización a través de receptores acopladas a proteínas G o a través de integrinas, como la αIIßb3, es dependiente de tirosina kinasas y de la activación de proteínas Ras asociada a la reorganización del citoesqueleto, secreción de los gránulos, activación del receptor para el fibrógeno, retracción del coágulo y, posiblemente, la expresión de actividad coagulante86,87. Ni los receptores acoplados a proteínas G ni las integrinas poseen actividad tirosina kinasa intrínseca, como ocurre con los receptores de factores de crecimiento. Se supone que la activación del receptor lleva a la activación de kinasas que fosforilan sustratos implicados en el reclutamiento de complejos de moléculas señalizadoras y la activación de proteínas Ras86.
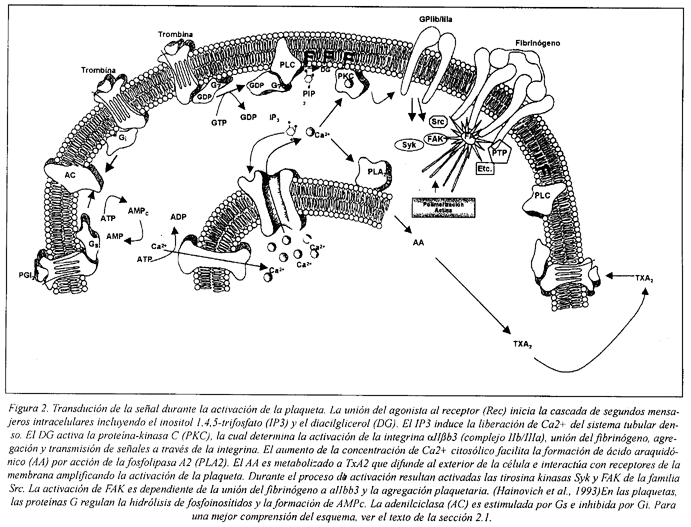
Los mecanismos de transdución de la señal implicados en la activación de la plaqueta se encuentran resumidos en el esquema de la Fig. 2.
2.1.1 Metabolismo de los fosfoinosítidos y fosfatos de inositol
Los agonistas que inducen secreción y agregación de las plaquetas estimulan la hidrólisis de fosfoinosítidos por la PLC. En las plaquetas y en otros tejidos se encuentra actividad PLC, tanto en las fracciones membranales como citosólicas, habiendo sido descritas ya varias formas de la enzima que se distinguen por su peso molecular, sustrato específico, dependencia de Ca2+, pH óptimo e inmunorreactividad. Las plaquetas poseen las formas: PLCß, reguladas por proteínas G, y PLCγ, reguladas por interacciones proteína-proteína que dependen del estado de fosforilación de las tirosinas23.
En las condiciones presentes en la plaqueta activada, la PLC hidroliza el fosfatidilinositol (PI), el fosfatidilinositol 4-fosfato (PIP) y el fosfatidilinositol 4,5-bifosfato (PIP2) en cantidades similares. Sin embargo, a las concentraciones de Ca2+ presentes en la plaqueta en reposo el PIP2 es el principal sustrato de la enzima67,88.
En las plaquetas en reposo, la interconversión de los fosfoinositidos ocurre lentamente, funcionando sólo para regular el contenido normal de estos compuestos en las membranas. El PI se encuentra tanto en la membrana plasmática como en las membranas del sistema tubular denso, mientras que el PIP y PIP2 existen, casi exclusivamente, en la membrana citoplasmática50.
Los fenómenos bioquímicos de la interconversión cíclica de los fosfoinositidos y fosfatos de inositol, han sido demostrados principalmente para plaquetas activadas con trombina. En este caso, se verifica una disminución transitoria de PIP2 acompañada por la formación de IP3 e IP2, seguida más tarde por una disminución de PI y acumulación de inositol monofosfato50,89,90.
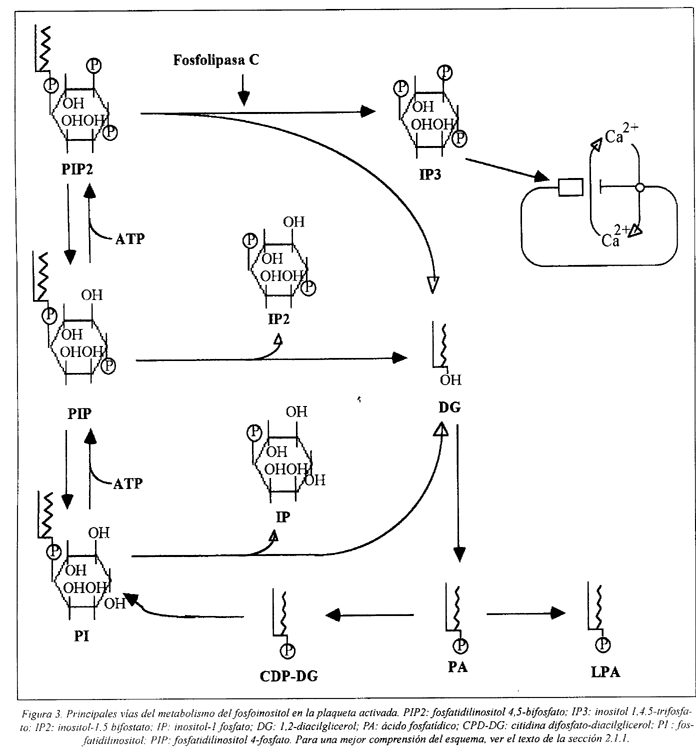
Las reacciones posteriores a la hidrólisis de PIP2 incluyen la conversión de DG en ácido fosfatídico (AF) y la regeneración de PIP2 por una serie de reacciones secuenciales catalizadas por transferasas y kinasas específicas, esquematizadas en la Fig. 3
El DG activa la proteína-kinasa C promoviendo su traslado del citoplasma hasta la membrana y aumentando la afinidad de la enzima para el Ca2+. La activación de la proteína-kinasa C requiere Ca2+ y fosfolípidos, siendo la fosfatidilserina un cofactor esencial. Esta kinasa cataliza la fosforilación de resíduos de serina y treonina en diversas proteínas implicadas en la secreción de los gránulos y en la exposición del sitio receptor para el fibrinógeno en el complejo GP IIb/IIIa. El principal substrato de la proteína-kinasa C es la proteína P47(47kD) o pleckstrina, cuya fosforilación ocurre de modo simultáneo a la liberación de serotonina de los gránulos densos. Otras proteínas constituyentes del citoesqueleto, por ejemplo la cadena ligera de la miosina (P20), la vinculina, la filamina, la talina y la caldesmon, son también sustratos de esta enzima64,65,77,82,91,92,93,94.
La activación de la proteína-kinasa C es esencial para que ocurra agregación y secreción, sin embargo existen proteínas que al fosforilarse inhiben las respuestas de la plaqueta. Es el caso de la IP3-fosfomonoesterasa que se activa y degrada el IP3, inhibiendo así los efectos de este segundo mensajero. Hay también evidencias de que la fosforilación de la proteína Gi, implicada en la transducción de la señal que inhibe la adenilciclasa, la inactiva promoviendo así el aumento del AMPc intracelular, y la consiguiente disminución del Ca2+ citosólico64,67,95,96. Estas señales inhibidoras pueden funcionar para atenuar y limitar la estimulación iniciada.
El DG se metaboliza rápidamente, principalmente, hasta ácido fosfatídico por acción de la diacilglicerol kinasa, y, en menor grado, se convierte en glicerol por diacilglicerol- y monoacilglicerol-lipasas. De esta última reacción puede resultar la liberación de ácido araquidónico, aunque la mayor parte de este ácido graso es liberado de los fosfolípidos de la membrana por la PLA250.
El IP3 promueve la liberación del Ca2+ secuestrado en el sistema tubular denso (análogo al retículo endoplasmático de otras células), llevando al aumento de la concentración citosólica de este catión. El mecanismo de movilización del Ca2+ implica la unión del IP3 a un receptor específico en la membrana del STD, que resulta en la activación de canales de Ca2+. Se supone que el ácido lisofosfatídico y el ácido fosfatídico tienen propiedades ionofóricas similares a IP3. La acción de IP3 es modulada por el pH, cationes monovalentes, otros cationes divalentes y GTP. El aumento del pH que acompaña la activación de las plaquetas, incrementa drásticamente la unión del IP3 a su receptor72,73,74,97.
La hidrólisis de PIP2 por la fosfolipasa C requiere una concentración mínima de Ca2+, pero no es regulada por concentraciones de Ca2+ por encima de ese nivel, al contrario de la PLA2. Sin embargo, la acción de la enzima sobre el PI es potenciada por concentraciones elevadas de Ca2+, lo que explica que la formación de DG exceda la de IP3 durante la activación de la plaqueta67.
El aumento de Ca2+ intracelular, per se, no es suficiente para determinar la respuesta completa de reacción de liberación; ésto solamente ocurre cuando la proteína-kinasa C es activada. Este hecho sugiere la importancia de la sinergia entre la fosforilación de proteínas inducida por la proteína-kinasa C y la movilización del Ca2+ 64,81,98.
La PLA2, activada por el aumento de la concentración de Ca2+, inicia el metabolismo del ácido araquidónico y consecuente formación de endoperóxidos de prostaglandinas y TXA2. Estos eicosanoides difunden al exterior de la plaqueta y activan la PLC, a través de la interacción con receptores específicos99.
2.1.2 Metabolismo del ácido araquidónico
Otra vía de transducción de la señal activada por la estimulación de la plaqueta mediada por un receptor, se inicia con la liberación del ácido araquidónico de fosfolípidos por acción de la fosfolipasa A2. Esta vía tiene un papel central en la activación plaquetaria inducida por los agonistas que no activan directamente la PLC y en la amplificación de la señal de activación.
La PL A2 puede ser activada por una proteína G transductora de la señal de unión de un agonista a su receptor específico. A la inversa de otras interacciones receptor-efector mediadas por proteínas G, la activación de la PLA2 parece ser mediada por las subunidades Gbγ en vez de la Gα 64,67,100. La activación de esta enzima también tiene lugar como consecuencia de un aumento de Ca2+ citoplasmático o del pH. Por ejemplo, la adrenalina puede estimular un intercambiador de Na+/H+ que lleva a un aumento del pH y subsiguiente activación de la fosfolipasa A2100,101. Las plaquetas poseen múltiples isoformas de estas enzimas, y se probó que la forma citoplasmática (cPLA2) es fosforilada por una MAP kinasa (p38MAPK), llevando a sugerir que el incremento de Ca2+ más la fosforilación eran responsables de la asociación de PLA2 con membranas donde se localizan sus sustratos. Sin embargo, estudios más recientes demostraron que la inhibición de p38MAPK no inhibe la liberación de ácido araquidónico, por lo que el mecanismo de regulación de PLA2 está aún indeterminado68. La PLA2 libera ácido araquidónico de la posición C2 de fosfolípidos de la membrana, sobre todo la fosfatidilcolina y la fosfatidiletanolamina67,100.
El ácido araquidónico puede ser metabolizado por dos vías: por la vía de la cicloxigenasa o por la vía de la lipoxigenasa. La vía más importante responsable de las reacciones de la plaqueta en la hemostasia es la que depende de la cicloxigenasa. Por la acción de esta enzima se forman dos endoperóxidos muy inestables, PGG2 y PGH2 que a su vez serán metabolizados a diferentes prostaglandinas. En la plaqueta, la PGH2 se transforma predominantemente en TxA2, por acción de la tromboxano-sintetasa (Fig. 4). Tanto los endoperóxidos PGG2 y PGH2 como el TxA2 son sustancias liberadas por la plaqueta durante su activación, siendo el TxA2 el más potente en la inducción de la activación plaquetaria. Estas sustancias se difunden al exterior de la célula, amplificando los fenómenos de activación por dos vías:
1) promoviendo la activación de la fosfolipasa C a través de la interacción con receptores específicos de la membrana citoplasmática, llevando a las respuestas intracelulares mediadas por el IP3 y DG.
2) induciendo la liberación de ADP de los gránulos densos y la liberación de fibrinógeno, vWF, y trombospondina de los gránulos a, sustancias que, en conjunto, están implicadas en la formación y estabilización de los agregados plaquetarios6,50,64,99.
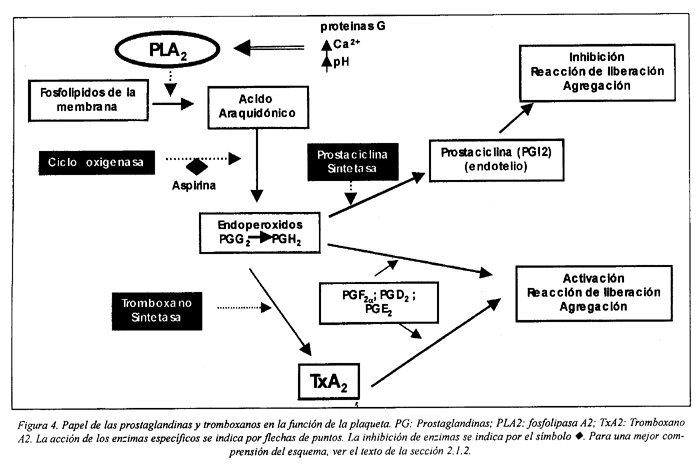
Las células endoteliales y del músculo liso pueden captar los endoperóxidos liberados por las plaquetas y sintetizar la PGI2, sustancia inhibidora. Por otra parte, las plaquetas pueden convertir el ácido araquidónico y leucotrienos derivados de neutrófilos en TXA2 o lipoxinas proinflamatorias, respectivamente97.
Estudios de fracciones celulares sugieren que la cicloxigenasa y tromboxano-sintetasa, enzimas requeridas para metabolizar el AA a TxA2, se encuentran localizadas en el sistema tubular denso. La membrana plasmática y el sistema tubular denso están interconectados y el ácido araquidónico liberado por la PLA2 puede difundir de un sistema de membrana al otro. No obstante, hay evidencias de que los acervos de AA que se disponibilizan más rápidamente están también localizados en el sistema tubular denso. Esto puede significar que el STD es el lugar preferencial de liberación del AA así como de su metabolismo64,67.
2.1.3 Tirosina Kinasas y proteínas Ras:
La transducción de la señal en las plaquetas implica tirosina kinasas y proteínas Ras, que regulan efectores apropiados en la respuesta celular.
En las plaquetas se encuentran muchas de las moléculas con dominios SH2, SH3 y PH identificadas en estudios de señalización a través de receptores de factores de crecimiento, y que son incorporadas en una variedad de complejos señalizadores con localizaciones subcelulares específicas. Por otra parte, las plaquetas contienen diferentes miembros de la familia Ras de moléculas señalizadoras, así como sus reguladores y enzimas efectoras que éstas regulan86.
Las moléculas Ras son proteínas de bajo peso molecular que unen GTP, poseen actividad GTPásica, y son activadas a través del ciclo entre los estados unida a GTP (activa) y unida a GDP (inactiva). Este ciclo es regulado por 2 clases de proteínas:
a) GEFs (Guanine Nucleotide Exchange Factors), proteínas que catalizan la disociación de GDP permitiendo la asociación de GTP y así activan la proteína.
b) GAPs (GTPase Activating Proteins), proteínas que inducen la hidrólisis de GTP, favoreciendo la forma inactiva de la proteína unida a GDP.
Los reguladores de Ras en las plaquetas incluyen el GEF, Sos y los adaptadores Grb2 y Shc, y el GAP p21Ras86.
Se ha observado que durante el proceso de activación plaquetaria son activadas tirosina kinasas citoplasmáticas, y estudios donde se usaron inhibidores de tirosina kinasas demostraron que la actividad tirosina kinasa es esencial en la transducción de la señal102.
Estas kinasas son activadas por receptores acoplados a proteínas G, o por receptores de adhesión como consecuencia de la unión del ligando. La transmisión de señales a través de la integrina a IIbß3 es el proceso mejor caracterizado. La ocupación de muchos receptores acoplados a proteínas G lleva a la rápida activación de tirosina kinasas incluyendo Src, Syk y PYK2. Aunque los mecanismos por los cuales las proteínas G se acoplan a las cascadas de tirosina kinasas no sean conocidos, el resultado es la fosforilación de innumerables proteínas, incluyendo PLCγ, Vav y cortactina (una proteína ligadora de actina cortical).
Muchas de las proteínas fosforiladas en residuos de tirosina, tras la activación de las plaquetas están implicadas en la activación de las proteínas Ras (por ejemplo p21rasGAP, Vav, Grb2, Shn) o en mediar los efectos de las proteínas Ras (p.ej. kinasas MAP Erk1 y Erk2)102.
Las proteínas de la superfamilia Ras determinan la activación de efectores incluyendo: MAP kinasas que fosforilan proteínas asociadas a los microtubulos y la PLA2; P65PAK que fosforila hsp27 asociada al citoesqueleto; PI-5 K que regula la polimerización de la actina; WASP implicada en la reorganización del citoesqueleto y cuya función se evidenció en el Síndrome de Wiskott-Aldrich (deficiencia de WASP) en el cual las plaquetas presentan morfología y organización del citoesqueleto anormales; p85/PI-3K implicada en la estabilización de las interacciones αIIßb3-ligando86.
Los agonistas que inducen la activación de la integrina αIIbß3 también contribuyen a la activación de Ras. La agregación está asociada a la activación de la tirosina kinasa p125FAK y constituye un ejemplo bien estudiado del ensamblaje de complejos de señalización asociado a las integrinas. La localización subcelular de FAK es determinada por una adhesión focal y por un sitio de unión a la integrina. Después de activada, FAK se autofosforila permitiendo la asociación de Src que, a su vez, promueve la fosforilación de residuos adicionales de tirosina, formando lugares de interacción con el adaptador Grb2. Además, FAK forma un complejo con los adaptadores p130 cas y paxilina y con GRAF, la proteína activadora de GTPasa de Rho87. Así, la activación de FAK requiere la ocupación de la integrina por el ligando, y la unión del agonista al receptor que lleva al incremento de Ca2+ y a la activación de PKC.23,66,87,103,104.
2.1.4 Formación del AMPc:
Los agentes que estimulan la adenilciclasa y aumentan los niveles de AMPc, tales como las prostaglandinas PGI2, PGE1 y PGD2, inhiben la función plaquetaria. Se supone que sus receptores están acoplados a Gs, responsable de la activación de la adenilciclasa.
La mayoría de los agonistas, a su vez, tienden a suprimir la formación de AMPc. Este efecto es mediado por Gi y se muestra fundamentalmente cuando la velocidad de formación de AMPc aumenta por encima de los niveles basales por la adición de estímulos tales como PGI266. Ejemplos de agonistas que inhiben la formación del AMPc son la trombina, la adrenalina y el ADP. Sin embargo, no todos los agonistas de las plaquetas consiguen este efecto. Ni el PAF, ni la vasopresina inhiben la formación de AMPc estimulada por PGI2 en las plaquetas intactas. Por consiguiente, sus receptores no están acoplados a Gi. Así, es posible disociar la supresión de la adenilciclasa de la activación de las plaquetas, lo que sugiere que una disminución en los niveles de AMPc plaquetario no es necesariamente la señal para la activación plaquetaria64,66.
A pesar de estar bien establecido que niveles elevados de AMPc están asociados a la inhibición de la función plaquetaria, ningún mecanismo único parece ser responsable del efecto inhibidor del AMPc. En general, se piensa que los efectos del AMPc son mediados por una proteína-kinasa dependiente de AMPc (PKA). Los sustratos plaquetarios de esta enzima incluyen la cadena ß de la glicoproteína Ib, la proteína ligadora de la actina, la kinasa de la cadena ligera de la miosina y una proteína recientemente descrita, VASP, que se localiza en los complejos integrina-citoesqueleto y regula la polimerización de actina87.
Se ha demostrado que agentes que aumentan los niveles de AMPc afectan a una variedad de eventos bioquímicos requeridos para la activación inducida por los agonistas. El AMPc provoca los siguientes efectos:
- disminuye la unión de la trombina
- disminuye la formación de DG e IP3 dependiente de la PLC
- inhibe la ciclo oxigenasa plaquetaria
- inhibe la señal DG para la activación de PKC incrementando su metabolismo a fosfoinositidos
- inhibe directamente la PKC
- inhibe respuestas como la polimerización de actina y expresión del receptor para el fibrinógeno.
El principal mecanismo por el cual el AMPc inhibe la activación de las plaquetas es su antagonismo en las respuestas mediadas por el Ca2+. El AMPc disminuye los incrementos transitorios del Ca2+ citosólico, inducidos por los agonistas, primeramente bloqueando la PLC. Adicionalmente, interfiere directamente en la liberación y recaptación de Ca2+ por el STD. La proteína kinasa dependiente del AMPc inhibe la liberación de Ca2+ mediada por el IP3 y, por otra parte, el AMPc acelera la entrada de Ca2+ al sistema tubular denso, estimulando el transportador Ca2+ATPasa105. A través de su efecto en la [Ca2+]i, el AMPc inhibe diversas respuestas incluyendo la liberación de ácido araquidónico mediado por la PLA2, aglomeración del citoesqueleto y la actividad de la cadena ligera de miosina97.
2.1.5 Formacion de GMPc
Otra importante vía inhibidora fisiológica de la plaqueta es la mediada por el incremento de GMPc citosólico. El vasodilatador endógeno óxido nítrico (o factor de relajamiento derivado del endotelio) difunde a través de la membrana plasmática y activa la forma soluble de guanilciclasa (citosólica) por un mecanismo independiente de receptores, determinando la síntesis de GMPc. Este segundo mensajero probablemente media en la inhibición de la plaqueta, activando la proteína kinasa dependiente de GMPc (PKG)97,106.
Los mecanismos moleculares por los cuales GMPc inhibe las plaquetas están menos claros. El GMPc tal como el AMPc bloquea la movilización de calcio, la activación de la fosfolipasa C y la liberación de ácido araquidónico mediado por la PLA2. Además, la fosforilación de VASP, relacionada con la inhibición de la agregación plaquetaria, también ocurre por acción de la PKG. Por otra parte, ha sido referido que el incremento de los niveles de GMPc retrasa la degradación de AMPc107.
2.1.6 Ca2+ citosólico
Cuando las plaquetas están en reposo, la membrana citoplasmática es relativamente impermeable al Ca2+, encontrándose éste en una concentración extracelular 10.000 veces superior a la intracelular. La concentración de Ca2+ intraplaquetario es del orden de los milimolares, aunque en el citosol sea apenas de 100 nM, lo que se traduce en una distribución intracelular no homogénea. La mayor parte del Ca2+ se encuentra almacenado en las siguientes estructuras celulares: mitocondrias, gránulos a, gránulos densos, cara interna de la membrana citoplasmática y, principalmente, en el sistema tubular denso44,108.
La activación de las plaquetas por la mayoría de los agonistas, probablemente por todos, está acompañada por un aumento de la concentración de Ca2+ libre citoplasmático ([Ca2+]i) que resulta, no solamente de la redistribución del calcio almacenado en las mencionadas estructuras, sino también de la difusión de Ca2+ extracelular hacia el interior de la célula a través de canales específicos para este catión. Aunque la magnitud y duración de las respuestas de [Ca2+]i a la activación aumenten en presencia de Ca2+ extracelular, los mecanismos por los cuales se genera la entrada de Ca2+ continúan sin esclarecer109. Los datos disponibles indican la existencia de canales de Ca2+ operados por receptores (ROC), los cuales pueden ser formados por subunidades de la proteína que funciona como receptor, o estar estrechamente unidos al receptor via proteina G110. Adicionalmente, parecen desempeñar un papel importante los canales regulados por los almacenes, en los cuales la entrada de Ca2+ es activada cuando los almacenes intracelulares están suficientemente vacíos111. Se ha sugerido que los complejos GP IIb/IIIa, al sufrir alteraciones conformacionales durante el proceso de activación, pueden funcionar como canales para el Ca2+, y además, proporcionar la exposición de canales específicos para este catión22,44,64.
El Ca2+ citosólico regula las respuestas celulares a través de diversas vías efectoras, incluyendo la activación de kinasas dependientes de Ca2+/calmodulina, proteasas dependientes de calcio, PLA2, PLC y proteína-kinasa C.
La cadena ligera de la miosina es el principal sustrato de la kinasa dependiente de Ca2+ calmodulina y la fosforilación de esta proteína está directamente implicada en el cambio de forma, contracción y secreción97. La fosforilasa kinasa también es dependiente de Ca2+-calmodulina y la activación de la fosforilasa lleva a un incremento del ATP citosólico necesario para la activación del aparato contráctil112.
Las proteasas dependientes de Ca2+ (Calpaina I y II) fragmentan la proteína ligadora de actina y talina, contribuyendo a la reorganización del citoesqueleto requerida para la agregación de las plaquetas. Adicionalmente, las calpaínas participan en la expresión de actividad procoagulante y en la formación de las micropartículas22,113.
El aumento de la concentración de Ca2+ lleva a la activación de las fosfolipasas C y A2, como se mencionó anteriormente, y algunos de los productos del metabolismo del ácido araquidónico a su vez aumentan el Ca2+ citosólico68.
La activación de las respuestas plaquetarias por el aumento de Ca2+ citosólico es un fenómeno gradual. La cantidad de Ca2+ requerida para la activación aumenta con cada paso secuencial en la activación de la plaqueta en un orden aproximado de: cambio de forma, agregación, secreción de los gránulos densos, secreción de los gránulos α, liberación de ácido araquidónico y secreción de los lisosomas50,114.
La liberación del calcio de los lugares de almacenamiento intracelulares es inducida por los segundos mensajeros IP3, PA y LPA, mientras que el AMPc parece ser el principal regulador de su acumulación en el sistema tubular denso, estimulando el transportador Mg2+/Ca2+ATPasa45,50,64,105. Adicionalmente, la activación de la 5-fosfomonoesterasa por la PKC, inactiva al IP3, como se mencionó anteriormente.
2.2. Respuestas funcionales
Las plaquetas pueden ser activadas por un gran número de sustancias, a las cuales reaccionan en pocos segundos. Cuando son expuestas a estos agonistas, las plaquetas presentan diferentes respuestas, que se encuentran íntimamente asociadas in vivo, pero que, in vitro, pueden ser estudiadas separadamente: adhesión y cambio de forma, agregación, secreción y expresión de actividad procoagulante64.
2.2.1. Adhesión:
La primera respuesta hemostática plaquetaria consiste en la adhesión de las plaquetas a la matriz subendotelial, que normalmente se expone después de una lesión vascular, en que hay descamación y/o ruptura del endotelio (Fig. 5).
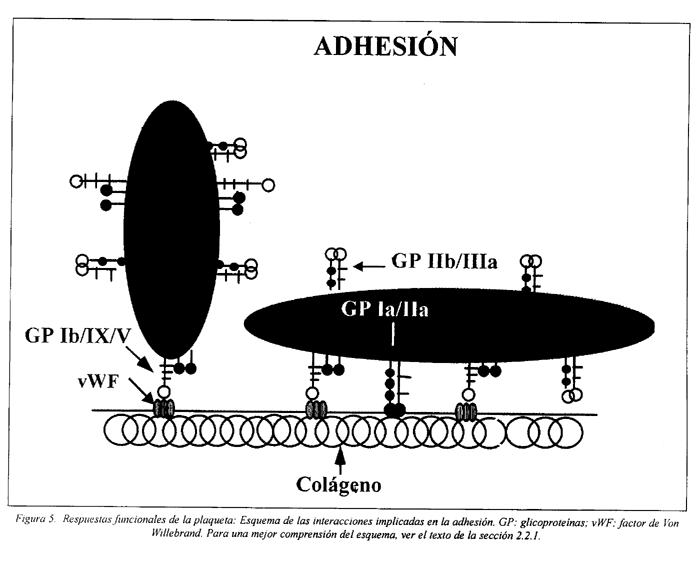
En regiones en que la circulación ocurre con bajo coeficiente de cizallamiento las plaquetas se adhieren al colágeno, fibronectina, laminina, vitronectina y trombospondina del subendotelio. Este proceso es mediado por receptores específicos de membrana, ya mencionados.
Bajo condiciones de alto coeficiente de cizallamiento, como se verifican en la mayor parte del territorio arterial y en la microcirculación, la interacción de las plaquetas con las sustancias de la matriz subendotelial, anteriormente mencionadas, requiere un cofactor adicional, el factor de vWF. El complejo GP Ib/IX/V es el principal receptor implicado en la adhesión en estas condiciones de flujo27,33,46,115,116,117.
La adhesión inicial de las plaquetas a un vaso lesionado es un proceso pasivo, pero esta unión resulta en la activación celular y esa adhesión es favorecida sobre todo por la consiguiente expansión de la plaqueta.
2.2.2 Cambio de forma
A la adhesión de las plaquetas le siguen diversas modificaciones de su forma, primero pasando de discoides a esferoides, siguiéndose la expansión con emisión de pseudópodos, como resultado de la reorganización del citoesqueleto (Fig. 6). Esto facilita, no solamente el proceso de adhesión de las plaquetas al subendotelio, sino también la interacción con otras plaquetas.
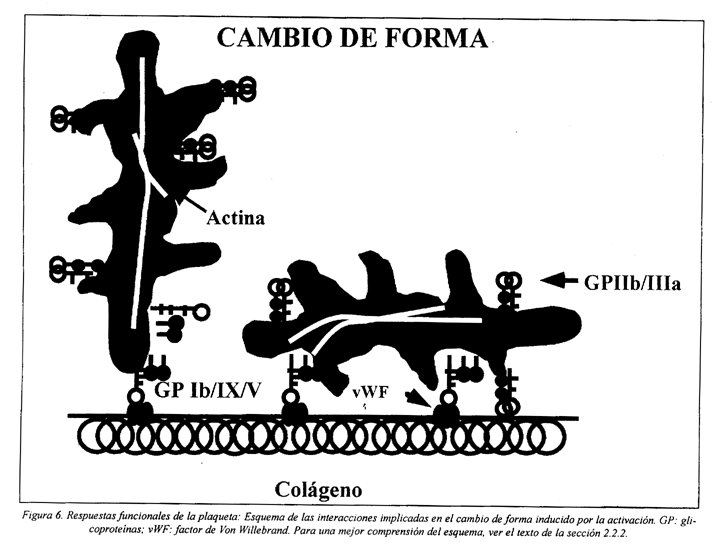
Durante la activación plaquetaria, mientras se dan los cambios de forma, ocurre la despolimerización reversible de los microtúbulos y su desplazamiento de la periferia a una posición más central97,118. Simultáneamente, se incrementa el contenido en actina filamentosa, proceso regulado por el incremento de la concentración de Ca2+ y por la interacción de proteínas reguladoras de la actina con lípidos de la membrana35,119,120. Por ejemplo, el PIP2 se une a la profilina y evita su interacción con la actina. Después de la hidrólisis del PIP2 por la PLC, la profilina es liberada de la membrana, interactúa con la actina G y ésta cambia ADP por ATP (actina G-ATP). La actina G-ATP tiene mayor afinidad para la extremidad del filamento, resultando en un aumento de la polimerización46,47.
La fosforilación de la miosina lleva a su incorporación en el citoesqueleto y formación de un gel contráctil que contribuye al cambio de forma y permite la centralización de los gránulos. La asociación de la actina polimerizada con la miosina es capaz de generar una fuerza que puede ser convertida en movimiento externo (extensión) o movimiento interno (contracción). Paralelamente, en los pseudópodos la actina polimerizada forma haces. Adicionalmente, los filamentos de actina originan contactos focales con la membrana y se desarrollan los sitios de adhesión focal46.
Los sitios de adhesión focal son dominios especializados de la membrana, donde la actina del citoesqueleto está anclada a integrinas, tales como la αIIbß3, a través de interacciones que implican a α-actinina, viculina y talina. Estas estructuras envuelven además, el ensamblaje de complejos de moléculas señalizadoras, incluyendo la tirosina kinasa p125FAK, y son reguladas por una proteína de la familia Ras. La proteína Rho activada estabiliza la adhesión focal, mientras que su inactivación lleva a la disrupción de las adhesiones focales. Las adhesiones focales suministran fuerza contráctil a través de la membrana y contribuyen al cambio de forma46,86.
Aunque la mayoría de los agentes agregantes induzca las dos fases del cambio de forma, la adrenalina induce sólo la formación de pseudópodos y la plaqueta mantiene su forma discoide. Este hecho sugiere, por un lado, que los dos procesos son independientes y, por otro lado, que el cambio de discoide a esferoide no es esencial para la agregación64.
En condiciones fisiológicas, el cambio de forma precede a la agregación y a la secreción. La agregación requiere la emisión de pseudópodos y la centralización de los gránulos, lo que ocurre durante la esferización de la plaqueta y que puede ser un prerrequisito para la secreción64.
2.2.3 Agregación
La agregación consiste en la unión de las plaquetas activadas unas a las otras. La activación, que ocurre después del contacto con el subendotelio, con agonistas liberados por otras plaquetas activadas o con trombina, inicia una serie de alteraciones que llevan a la expresión de receptores funcionales de la membrana, los cuales determinan la interacción plaquetaplaqueta (Fig. 7).
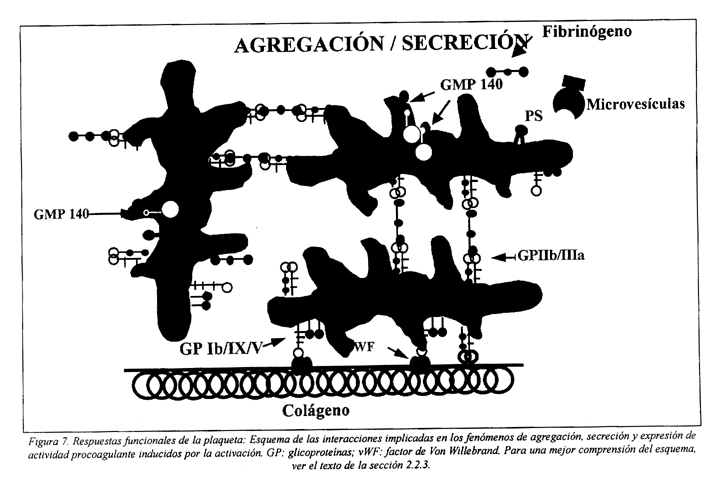
Este proceso es dependiente de Ca2+ extracelular y del fibrinógeno, que forma un puente entre las plaquetas. En la fase inicial de la agregación la unión del fibrinógeno a su receptor, el complejo GP IIb/IIIa (αIIbß3 ) es reversible, pero enseguida se convierte en irreversible. Esta irreversibilidad se debe a la estabilización de los puentes de fibrinógeno por varios factores: alteraciones intrínsecas del receptor inducidas por el ligando; aglomeración de complejos; internalización del receptor; y la unión al fibrinógeno y/o αIIbß3 de la trombospondina u otras proteínas adhesivas liberadas por los gránulos α64,87,121.
Otras proteínas adhesivas que contienen la secuencia clásica de reconocimiento de integrinas Arg-Gly-Asp, pueden interactuar con αIIbß3, pero el ligando preferencial en la agregación es el fibrinógeno. Sin embargo, si hubiera una deficiencia en este factor, el vWF puede formar los puentes entre las plaquetas122, y en condiciones en que las fuerzas de cizallamiento son muy elevadas el factor de vWF parece ser el principal cofactor de la agregación123.
De las numerosas sustancias capaces de inducir la agregación plaquetaria in vitro podemos distinguir:
1) agentes agregantes primarios: aquellos que llevan directamente a la agregación por mecanismos no mediados por la síntesis de prostanoides o por la liberación del contenido plaquetario de ADP, o sea, exponen directamente los sitios receptores del fibrinógeno, de los que son ejemplo la trombina y el ADP.
2) agentes agregantes secundarios: sustancias que promueven la agregación plaquetaria al estimular la liberación de ADP y/o producción de endoperóxidos y TXA2.
En cualquier caso la interacción de las plaquetas como consecuencia de la agregación induce reacción de liberación. Por este motivo, la agregación llevada a cabo por los agentes agregantes primarios en ciertas circunstancias es bifásica, o sea, una agregación inicial directa y una agregación subsiguiente, asociadas a la reacción de liberación, pero no necesariamente consecuencia de ésta64.
En el proceso de agregación aparecen por lo menos dos mecanismos de amplificación por retroalimentación positiva, la liberación de ADP, y la síntesis de prostanoides, particularmente TXA2, PGG2 y PGH2 que, al unirse a receptores de la membrana plaquetaria, estimulan la hidrólisis de fosfatidilinositol, la movilización de Ca2+ y fosforilación de proteínas, y la exposición de receptores del fibrinógeno. Ante la presencia de estímulos fuertes, tales como el colágeno y la trombina, puede haber un tercer factor amplificador, que consiste en la síntesis y liberación de PAF. Este factor también se une a un receptor específico de la membrana, activa el ciclo del fosfatidilinositol y lleva a la exposición de lugares de unión del fibrinógeno64.
La agregación determina transmisión de señales al interior a través de αIIbß3 implicados en la estabilización de grandes agregados, expansión de las plaquetas, secreción de los gránulos, retracción del coágulo y, posiblemente, en la expresión de actividad procoagulante.
2.2.4 Secreción
Cuando las plaquetas son activadas se produce la secreción de numerosas sustancias contenidas en sus gránulos. Los gránulos densos segregan, entre otras sustancias, ADP y serotonina que van a activar otras plaquetas, que se encuentren en las proximidades; los gránulos a liberan proteínas adhesivas tales como vWF, trombospondina, fibronectina y fibrinógeno, que se concentran en la superficie de la plaqueta, además de proteínas específicas de la plaqueta, como es el caso del PF4 y de la ß-TG. Los lisosomas liberan principalmente hidrolasas ácidas.
La secreción de los gránulos densos y de los gránulos α, ocurre después de la estimulación con todos los agonistas, aunque la liberación de los gránulos α se observe a concentraciones de agonista relativamente superiores. La secreción de los lisosomas requiere estímulos muy fuertes, y se observa después de la estimulación con altas concentraciones de trombina y colágeno. Además, la secreción de los lisosomas es lenta e incompleta, hasta un máximo de 60% de los gránulos, mientras que los gránulos densos y a segregan aproximadamente 100% de sus contenidos entre 1 y 2 minutos124.
La secreción está determinada por los segundos mensajeros sintetizados en respuesta a la activación plaquetaria. Los agonistas fuertes tales como el colágeno y la trombina pueden inducir secreción sin agregación concomitante2.
El ensamblaje y contracción del sistema actina-miosina es responsable de la centralización y la secreción activa del contenido granular. Una vez centralizados se establece una comunicación entre los gránulos y el sistema canalicular abierto.
Durante el proceso de secreción, las membranas de los gránulos se funden con las del sistema canalicular abierto y de esta forma, proteínas de las membranas de los gránulos quedan expuestas en la membrana plasmática. Los gránulos α contribuyen con la expresión de dos glicoproteínas específicas, la GMP140 (Granule Membrane Protein), también conocida por P-selectina o PADGEM (Platelet Activation-Dependent Granule-External Membrane), y la GMP-33, e incluso con una cantidad adicional de complejo GP IIb/IIIa y de GP IV. La presencia de GP IIb/IIIa en este local específico parece sugerir que este receptor facilita la expresión de las proteínas adhesivas de los gránulos a que aparecen unidas a la superficie durante la activación de la plaqueta9,120,125,126,127.
La presencia de GMP140 en la superficie de la membrana cambia las propiedades funcionales de la plaqueta, mediando la interacción de las plaquetas activadas con neutrofilos, monocitos, y algunos subtipos de células T, un proceso que puede ser importante en la inflamación y la trombosis 9,120,128,129,130,131.
Otras proteínas, cuya función no está todavía esclarecida, se expresan en la membrana plasmática después de la activación. Es el caso de una proteína de los gránulos densos, de peso molecular 40.000, y de las glicoproteínas de la membrana lisosomal GP53 o LIMP-CD63 (Lisosomal Integral Membrane Protein), LAMP-1 y LAMP-28,124,132.
2.2.5 Expresión de actividad procoagulante y formación de micropartículas
Además de constituir los elementos básicos del trombo hemostático, las plaquetas activadas proporcionan una superficie que permite la asociación de complejos enzimáticos responsables de la generación de trombina. El complejo enzimático que convierte la protrombina en trombina, designado como protrombinasa, está compuesto por el factor V activado (Va), factor X activado (Xa), iones Ca2+ y protrombina y estos agentes interactúan en la superficie de vesículas lipídicas que contienen fosfolípidos aniónicos. La actividad procoagulante de las plaquetas activadas, inicialmente designada factor plaquetario 3 (PF3), es debida a la exposición de fosfolípidos aniónicos en la capa externa de la membrana, especialmente la fosfatidilserina, permitiendo la asociación de los componentes del complejo protrombinasa2,3,133,134,135,136.
Adicionalmente, las plaquetas contribuyen a la formación del complejo protrombinasa, liberando el acervo de factor V que se encuentra en los gránulos α y que corresponde aproximadamente al 20% del factor V existente en la sangre. El factor Xa forma un complejo equimolar con el factor V unido a la plaqueta, y también se une a los fosfolípidos de la membrana, a través de resíduos amino-terminales de ácido γ-carboxilglutámico e iones Ca2+ 3,137.
De modo similar, la reacción que genera Xa a partir de X, que implica el factor IX activado (IXa) y el factor VIII, puede ocurrir en la superficie de la plaqueta, lo que hace de la membrana plaquetaria activada una superficie altamente trombogénica2,3,137.
En la membrana de la plaqueta en reposo la distribución de los fosfolípidos es asimétrica, estando la fosfatidilserina y la fosfatidilcolina concentradas en la capa interna. La estimulación de las plaquetas por agonistas fisiológicos, como el colágeno o la trombina, determina la pérdida de esta asimetría y la exposición de fosfatidilserina (PS) en la capa externa, proceso que es acompañado por el desprendimiento de microvesículas membranales de la superficie (Fig. 7).
La asimetría de los fosfolípidos está regulada por las actividades cooperativas de tres transportadores:
a) la translocasa de aminofosfolípidos dependiente de ATP, la cual rápidamente transporta PS y PE de la capa externa hacia la capa interna.
b) la flopasa lipídica inespecifica dependiente de ATP, la cual transporta lípidos de la capa interna a la capa externa.
c) la scramblasa lipídica no específica, dependiente de Ca2+, la cual permite que los lípidos se muevan al azar entre las dos capas.
A [Ca2+]i fisiológicas, se promueve la asimetría de PS porque la translocasa y la flopasa son activas, pero la scramblasa es inactiva. Tras la activación celular, el incremento de [Ca2+]i induce una distribución al azar de los PL en la membrana, ya que inhibe la translocasa y estimula la actividad scramblasa. La [Ca2+]i aumentada puede también resultar en la activación de la calpaína, la cual facilita el burbujeo (blebbing) de la membrana y la liberación de microvesículas que expresan PS138.
El desprendimiento de las microvesículas está relacionado con la disrupción del citoesqueleto de la membrana plaquetaria, especialmente la hidrólisis de la ABP mediada por la calpaína y la disrupción de la asociación de los filamentos de actina con la superficie de la membrana139,140. Estas microvesículas, o micropartículas, contienen la mayor parte de la actividad atribuida a las plaquetas.
2.3. Agonistas plaquetarios y sus receptores
Los agonistas fisiológicos que pueden activar las plaquetas son muy variados e incluyen trombina, TXA2, PAF, ADP y adrenalina.
Hasta hace poco se conocía más de las respuestas plaquetarias a diversos agonistas que sobre la estructura de sus receptores. Sin embargo, en los últimos años se han hecho progresos en esta área, habiéndose clonado con éxito los receptores de la trombina, TXA2, PAF y ADP141. Todos los receptores de agonistas que interactúan con proteínas G, identificados hasta al momento, consisten típicamente en una cadena polipeptídica con una porción N-terminal extracelular, una porción C-terminal intracelular y siete dominios transmembrana.
2.3.1 Trombina
La trombina es una proteasa de serina, presente en el plasma en forma del precursor inactivo protrombina, que desempeña un papel central en el mecanismo de la coagulación al convertir el fibrinógeno soluble en monómeros de fibrina que polimerizan espontáneamente formando fibras. Adicionalmente, la trombina es un agonista fuerte e importante activador de las plaquetas in vivo.
Cuando se añade a las plaqueta in vitro, la trombina causa cambio de forma, agregación y secreción de los gránulos densos, gránulos α y lisosomas. Las plaquetas contienen aproximadamente 2.000 copias del receptor específico de la trombina, y éste se parece a otros que interactúan con proteínas G. La trombina hidroliza el receptor entre los residuos Arg-41 y Ser-42, y se expone un peptido ligando con la secuencia SFLLR, el cual se une a una porción específica del receptor, activándolo. Todas las respuestas de la plaqueta requieren la trombina proteolíticamente activa. Los péptidos sintéticos cuya secuencia comience por SFLLR, son capaces de mimetizar los efectos de la trombina en la plaqueta y son conocidos por TRAPs (Trombin-Receptor-Activanting-Peptides) 142,143,144.
Dependiendo de la concentración, la trombina activa múltiples vías de transducción de la señal incluyendo:
a) activación de diversas formas de PLC, con la resultante formación de IP3 y DG.
b) inhibición de la adenilciclasa.
c) activación de tirosina kinasas y proteínas Ras.
d) activación de PLA2.
Estas respuestas pueden ser detectadas a concentraciones tan bajas como 0,1 nM. La activación plaquetaria inducida por la trombina no es dependiente de la formación del TXA2, aunque este metabolito contribuya a las respuestas observadas. La activación de plaquetas tratadas con aspirina necesita concentraciones aumentadas de trombina para superar el bloqueo del fármaco68. El receptor GP Ib/IX/V también funciona como lugar de unión de alta afinidad para la trombina, participando en la propagación de la respuesta a este agonista en la activación y agregación plaquetaria31,34,145.
2.3.2 ADP
El ADP se encuentra almacenado en los gránulos densos de las plaquetas, de los cuales se libera tras la activación plaquetaria.
La exposición de las plaquetas al ADP produce un incremento de [Ca2+]i, rápido influjo de Ca2+, activación (débil) de la PLC e inhibición de la adenilciclasa. El ADP causa cambio de forma, activación del receptor del fibrinógeno, agregación y secreción. Se produce, también TXA2 que convierte la agregación reversible en agregación irreversible, la cual es referida como la segunda onda de agregación.
Las respuestas plaquetarias inducidas por el ADP fueron inicialmente atribuidas a un único receptor del tipo purinérgico P2T 146. Sin embargo, estudios más recientes pusieron de manifiesto la posibilidad de que los efectos del ADP en las plaquetas sean mediados por 3 subtipos de receptores P2 147,148,149. Según Kunapuli y Daniel147, un receptor está acoplado por Gi a la inhibición de la adenilciclasa (P2TAC), otro acoplado por Gq a la activación de PLC (P2Y1) y el tercero, es un receptor ionotrópico P2X1 mediador del influjo rápido de Ca2+. El receptor P2Y1 media en el cambio de forma inducido por el ADP, mientras que el receptor P2TAC es esencial para la agregación, aunque esta respuesta resulte de la señalización a través de P2TAC y P2Y1150,151. Se conocen ya la estructuras moleculares de P2X1 y P2Y1, pero el receptor P2TAC no ha sido clonado todavía147.
Las tienopiridinas, ticlopidina y clopidogrel, cuando se administran in vivo, son supuestamente convertidas en el metabolito activo, el cual anula la inhibición de la adenilciclasa inducida por el ADP152.
2.3.3 Tromboxano A2 y endoperóxidos PGG2 y PGH2
La plaqueta activada metaboliza el ácido araquidónico a TXA2 vía endoperóxidos de prostaglandinas PGH2 y PGG2. El TXA2 es un potente agonista plaquetario que amplifica las señales de activación. Tanto el TXA2 como los endoperóxidos son muy inestables, con vida medias inferiores a 5 minutos (TXA2 1min, PGH2 5min), y por eso se sintetizaron varios análogos, que permitieron estudiar los efectos de estas sustancias in vitro. Cuando se adicionan a las plaquetas, los agonistas TXA2/endoperóxidos causan cambio de forma, agregación y secreción. El TXA2 se considera un agonista fuerte porque estimula directamente la PLC. Los receptores para el TXA2/PGH2 están acoplados a la activación de PLC por la proteína Gq, mediando la hidrólisis de fosfoinositidos, el incremento de Ca2+ y fosforilación de proteínas. Estos agonistas no provocan la inhibición de la adenilciclasa68,97,153.
2.3.4 PAF
El factor activador de las plaquetas (PAF) es una molécula generada por diversas células circulantes activadas, incluyendo las plaquetas y los leucocitos. Es un lípido complejo (1-O-alquil-2-Oacetil-sn-glicerol-3-fosforilcolina) formado, a partir de un fosfolípido membranar, por la acción consecutiva de la PLA2 y una acil transferasa. Esta molécula es un potente activador de neutrófilos y un agonista fuerte de la activación plaquetaria, produciendo cambio de forma, agregación y secreción de las plaquetas. El PAF actúa mediante receptores especificos acoplados a la PLC, induciendo por eso el metabolismo de fosfoinositidos y movilización del Ca2+. Tal como el TXA2, PAF no inhibe la adenilciclasa. La importancia fisiológica de este agonista en la formación de trombos plaquetarios no está perfectamente esclarecida97,154.
2.3.5 Adrenalina
Las catecolaminas, liberadas a consecuencia de la activación fisiológica o patológica del sistema simpáticoadrenal, son importantes agonistas plaquetarios, especialmente la adrenalina.
Esta hormona (o neurotransmisor) potencia el efecto de otros agonistas plaquetarios y, a concentraciones elevadas, inicia respuestas plaquetarias incluyendo agregación, producción de TXA2 y secreción. Las plaquetas humanas poseen receptores adrenérgicos α2 y ß2, predominando los adrenérgicos α2 (presentes 200-300 lugares de unión/plaqueta)155. Las respuestas de activación plaquetarias son mediadas por los receptores adrenérgicos α2, mientras que los ß2 inducen inhibición y están acoplados a la estimulación de la adeniciclasa. La estimulación de receptores adrenérgicos α2 determina la inhibición de la adenil ciclasa vía proteína Gi, pero la transmisión de la señal por esta vía no es suficiente para provocar agregación. Este hecho llevó a proponer la existencia de dos receptores α2, uno inhibidor de la AC y otro implicado en la agregación. Recientemente, se ha demostrado que el receptor adrenérgico α2A activa la PKC a través de una vía independiente de la PLC y de la formación de DAG. Esta estimulación resulta en la activación del intercambiador de Na+/H+, la exposición de lugares de unión para el fibrinógeno en el complejo GPIIbIIIa y la agregación. La activación de PLC, cuando ocurre, es dependiente de la formación de TXA2.
Después de la ocupación prolongada de los receptores α2 por la adrenalina, se observa una disminución de las respuestas subsiguientes de agregación a la adrenalina, no estando aún claros los mecanismos que subyacen a esta insensibilización153.
Los efectos potenciadores de la adrenalina en la activación plaquetaria son un fenómeno extremamente relevante: se demostró que las concentraciones de adrenalina alcanzadas in vivo, tanto en condiciones fisiológicas como patológicas, aún siendo subagregantes per se, pueden inducir activación plaquetaria, interactuando con otros agonistas fisiológicos. Por otra parte, la adrenalina inhibe o revierte el efecto inhibidor de agentes antiagregantes, como la PGI2 o incluso la aspirina155.
3. Implicaciones de las plaquetas en procesos fisiológicos y patológicos
3.1. Procesos fisiológicos
3.1.1. Hemostasia y fibrinólisis
La principal función de las plaquetas es su participación en la hemostasia. Como hemos mencionado anteriormente, cuando ocurre una lesión de un vaso, las plaquetas se adhieren a las capas subendoteliales expuestas, son activadas y forman agregados plaquetarios, proceso amplificado por la liberación de sustancias proagregantes. Además de constituir una parte fundamental del trombo hemostático, las plaquetas activadas liberan sustancias procoagulantes y exponen en su superficie fosfolípidos que permiten el ensamblaje del complejo X-asa y del complejo protrombinasa, responsables de la activación del factor X y de la protombina, respectivamente. La trombina generada, a su vez, convierte el fibrinógeno soluble en la fibrina insoluble del coágulo sanguíneo. La retracción del coágulo se produce por fuerzas generadas a través de la contracción del sistema acto-miosina plaquetario. Esta fuerza es transmitida a través del complejo GPIIbIIIa, que interactúa con las extremidades de las mallas de fibrina.
Los mecanismos procoagulantes iniciados deben ser controlados, y la membrana plaquetaria posee función anticoagulante. Las plaquetas interactúan con la proteína C activada, la cual inactiva los factores Va y VIIIa156.
Las plaquetas también participan en la regulación de la fibrinólisis, el proceso que permite la lisis de la fibrina y la disrupción del coágulo. Durante la secreción son liberados inhibidores de la fibrinólisis, concretamente la α2-antiplasmina, y el inhibidor de tipo 1 del activador del plasminógeno (PAI-1), los cuales retrasan la destrucción del coágulo por las enzimas fibrinolíticas. Por otro lado, la plaqueta activada libera plasminógeno y suministra una superficie que favorece la generación de plasmina por la acción del activador tisular del plasminógeno (t-PA).
3.1.2. Función de soporte endotelial
Las plaquetas están implicadas en el mantenimiento y/o restablecimiento de la integridad de la pared del vaso, contribuyendo al proceso de reendotelización en los lugares de lesión.
Cuando ocurre una lesión de un tejido, las plaquetas inician su cicatrización y reparación. Este proceso requiere la activación de las plaquetas y la consiguiente liberación del contenido de sus gránulos, sobre todo de los gránulos α, los cuales almacenan sustancias estimuladoras del crecimiento y función de los fibroblastos y células endoteliales.
Los gránulos α contienen los factores de crecimiento PDGF y EGF que promueven el crecimiento y la mitosis del fibroblasto. Estas sustancias también poseen actividad quimiotáctica, principalmente el PDGF. Otro factor de crecimiento de los gránulos a implicado en este proceso es el TGF-ß, que a concentraciones bajas estimula la proliferación de los fibroblastos, mientras que a concentraciones elevadas lo inhibe. Adicionalmente, el TGF-ß estimula la secreción de proteínas por el fibroblasto, principalmente colágeno (de tipo I y II) y fibronectina. Normalmente la fibrosis tisular es acompañada de la proliferación del endotelio, porque tanto el TGF-ß como el EGF son angiogénicos. Las plaquetas contienen además, una sustancia citoplasmática específica para la proliferación del endotelio, designada como factor de crecimiento de células endoteliales derivado de la plaqueta (PD-ECGF)157.
Por otra parte, el PF4 de origen plaquetario contribuye a la regulación de la cantidad de fibras extracelulares, inhibiendo las colagenasas, proteasas fisiológicas responsables de la degradación de la matriz colagénica.
3.1.3. Función destoxificante
Las plaquetas pueden recaptar diversas sustancias a partir del plasma y transportarlas, participando así en procesos de destoxificación. Un ejemplo de esta función es la recaptación y transporte de la serotonina (5HT) de los lugares de síntesis y liberación a áreas de actuación o degradación metabólica. La serotonina es un potente vasoconstrictor, y está implicada en la hemodinámica cardiovascular y otros procesos1.
3.2. Procesos patológicos
3.2.1 Estados inflamatorios
La inflamación es un proceso multifactorial que envuelve diferentes tipos de células, incluyendo las plaquetas. Las plaquetas son activadas por diversos mediadores de la inflamación e inmunidad y, por otro lado, cuando son activadas, interactúan con leucocitos y liberan sustancias que actúan sobre estas células defensivas, o directamente sobre los agentes patógenos157.
La actuación directa en los agentes patógenos incluye la liberación de sustancias quimiotácticas y la fagocitosis seguida de bacteriólisis por los lisosomas.
El proceso inflamatorio determina la activación de las plaquetas a través de diversos mecanismos: interacciones con los fragmentos Fc de las IgGs, con inmunocomplejos de la fracción C1q del complemento y con antígenos plaquetarios específicos; o con mediadores de la inflamación liberados por diferentes células158.
El complejo C5b-9 del complemento actúa sobre un receptor específico de la plaqueta, siendo particularmente activo en determinar la expresión de actividad procoagulante y formación de micropartículas159.
Los monocitos, granulocitos y células endoteliales producen PAF. Por otro lado, los granulocitos liberan peróxido de hidrógeno, anión superóxido, mieloperoxidasa, neutrofilina, Catepsina G y elastasa, así como ácido araquidónico y algunos de sus metabolitos (PGs y TxA2)160.
Adicionalmente, también contribuye a la activación plaquetaria la generación de trombina, potenciada por la actividad procoagulante de los leucocitos, y la matriz subendotelial expuesta como consecuencia de la lesión vascular.
Finalmente, las plaquetas pueden ser activadas por los propios agentes infecciosos, virus, bacterias o sus toxinas. Por ejemplo las toxinas de tipo lipopolisacárido, pueden causar agregación plaquetaria, e incluso llevar a una situación de trombocitopenia. Algunos de estos agentes inducen la expresión de actividad procoagulante y este fenómeno puede ser el responsable de la coagulación intravascular diseminada asociada a las bacteriemias por gram negativos160.
Las plaquetas activadas, a su vez, liberan diversos factores que participan en la inflamación incluyendo: sustancias bactericidas, como la ß-lisina, sustancias vasoactivas que incrementan directamente la permeabilidad vascular (PGE2, HETE, proteínas catiónicas, serotonina e histamina) o indirectamente promoviendo la degranulación de los mastocitos, con la consecuente liberación de histamina, factores quimiotácticos y otros mediadores161.
El PAF, también liberado por las plaquetas, es quimiotáctico y proagregante para neutrófilos y monocitos. En estas células determina también la producción de anión superóxido, facilita la síntesis de leucotrienos en los neutrófilos, la producción de citokinas en los monocitos y estimula su capacidad citotóxica. A nivel de los leucocitos disminuye su proliferación y producción de IL-2 y aumenta su capacidad supresora y Natural Killer154.
Las plaquetas también contienen IL-1 que interactuando con el TNF, estimula fibroblastos, células del músculo liso y células endoteliales, induce la secreción de otros mediadores de la inflamación y aumenta la expresión de moléculas de adhesión y la actividad procoagulante162.
Otra proteína de origen plaquetaria, el PF4 es quimiotáctica para los neutrófilos y monocitos estimulando sus funciones antibacterianas. Por otra parte, la ß-TG es convertida extracelularmente por la catepsina G en el péptido activante de neutrofilos 2 (NAP2)163.
Las plaquetas activadas se adhieren a neutrófilos y monocitos. Esta adhesión es mediada por la P-selectina de las plaquetas y el ligando PSGL-1(ligando 1 de la glicoproteina P-selectina) presente en los neutrófilos y otros leucocitos, y participa en importantes interacciones funcionales entre leucocitos y plaquetas. El contacto membrana-membrana facilita el intercambio de mediadores solubles y, por otra parte, la adhesión per se puede funcionar como una señal estimuladora. Se demostró que la plaqueta activada, a través de la P-selectina, induce la generación de anión superóxido en los PMN (leucocitos polimorfonucleares) adherentes o la expresión del factor tisular en los monocitos164.
La interacción PMN-plaqueta permite la cooperación célula-célula en el metabolismo del ácido araquidónico(AA), ejemplo de metabolismo transcelular. En este proceso, una célula receptora usa intermediarios de una célula dadora para producir un nuevo metabolito, que puede tener una función totalmente diferente del metabolito formado por la célula dadora a partir de los mismos precursores. El metabolismo transcelular (PMN-plaqueta) del AA es dependiente de la P-selectina y resulta en la producción de TxB2 y LTC4163.
3.2.2 Trombosis y patologías vasculares
Las disfunciones arteriales oclusivas, incluyendo las enfermedades coronaria, cerebral y arterial periférica, constituyen la principal causa de morbilidad y mortalidad. En estas enfermedades arteriales subyacen tres procesos patológicos generales en los cuales las plaquetas están implicadas165:
1) la aterosclerosis, que se desarrolla a lo largo de varios años.
2) la trombosis, que se desarrolla rápidamente en lesiones ateroscleróticas dañadas es la principal causa de la oclusión vascular aguda e infartos.
3) vasoespasmo, que contribuye a la oclusión vascular
Aunque las plaquetas juegan invariablemente un papel en todos los fenómenos trombóticos, su mayor participación es a nivel arterial, donde el coeficiente de cizallamiento es elevado y las lesiones endotelilales frecuentes.
La patogénesis de la trombosis arterial ha sido conceptualizada como una forma exagerada de la hemostasia. Cuando ocurre una lesión vascular, las plaquetas se adhieren a la matriz subendotelial expuesta, son activadas y responden con una sucesión rápida de reacciones bioquímicas y cambios celulares, llevando al reclutamiento y agregación de plaquetas adicionales. Las plaquetas participan en la formación del trombo en la pared arterial dañada, particularmente en la pared arterial aterosclerótica por procesos celulares y bioquímicos similares. Las plaquetas adhieren, se expanden y se agregan en la pared lesionada. Adicionalmente, la liberación de una serie de sustancias vasoactivas, mitogénicas y procoagulantes; causa vasoconstricción, migración y proliferación de las células del músculo liso y formación de fibrina. Sin embargo, en contraste con la formación del tampón hemostático en un vaso sano, que es un proceso ordenado y regulado, la formación de trombos en las complejas lesiones ateroscleróticas puede ser incontrolada e impredecible165.
La activación plaquetaria está íntimamente ligada a la coagulación, ya que la superficie de las plaquetas activadas permite la activación del factor X y generación de trombina3. La trombina, a su vez, recluta nuevas plaquetas y amplifica la formación de agregados plaquetarios.
Los mecanismos de defensa que previenen la trombosis pueden ser superados por daños vasculares, alteración del flujo sanguíneo y perturbación de las moléculas tromborreguladoras, asociados a la superficie o en fase fluida, para limitar la respuesta protrombótica. Los tromboreguladores limitan o revierten la acumulación de plaquetas y la activación de la coagulación166.
En los últimos años se ha demostrado que la trombosis vascular es un proceso en que intervienen múltiples células, participando directamente no sólo las plaquetas, sino también otras células como las células endoteliales y los neutrófilos107.
Los fenómenos trombóticos son desencadenados en las lesiones vasculares patológicas, como las fisuras de placas ateroscleróticas, donde hay condiciones que conducen a la respuesta inflamatoria. Por otra parte, las lesiones ateroscleróticas llevan a un estrechamiento de los vasos, creando elevadas tensiones de cizallamiento (shear stress) que promueven la acumulación de plaquetas e interacciones entre diversos tipos de células tanto en la fase líquida como en la pared del vaso. Las fuerzas de cizallamiento superiores a 50 dinas/cm2 inducen adhesión, agregación y secreción de las plaquetas. La adhesión es dependiente del vWF que forma puentes entre la GPIb y el subendotelio. La agregación, en estas condiciones es mediada también por el vWF que interactúa con GPIb o (en menor extensión) con GPIIbIIIa, pero no por el fibrinógeno plasmático167. Los mecanismos moleculares por los cuales las fuerzas de cizallamiento transmiten la señal de estimulación a través de la membrana, no son conocidos pero parecen ser iniciados por la GPIb. Estos ambientes protrombóticos son agonistas más fuertes que aquellos encontrados en la superficie de vasos sanos con trauma166,168.
La activación de las plaquetas a consecuencia de la lesión de un vaso provoca una respuesta en las células endoteliales, la cual limita o revierte las consecuencias oclusivas de la acumulación de plaquetas. Si las fuerzas agonistas son fuertes, la acumulación de las plaquetas puede tornarse irreversible.
Se ha demostrado que las plaquetas son inhibidas en las proximidades de la célula endotelial y esta inhibición es debida por lo menos a 3 sistemas tromborreguladores de la célula endotelial107:
1) producción de eicosanoides, particularmente la PGI2.
2) producción del factor relajante derivado de la célula endotelial (EDRF) u óxido nítrico.
3) presencia de una ecto-nucleosidasa, que metaboliza el ADP liberado.
Las plaquetas participan directamente en la síntesis de PGI2 por las células endoteliales, liberando ácido araquidónico y endoperóxidos. Estos compuestos son capaces de servir como precursores para la síntesis de PGI2107.
La deposición de las plaquetas en el endotelio no funcional puede precipitar los síndromes coronarios agudos, porque se produce un desequilibrio entre los elementos vasoconstrictores (se rotonina, TxA2) y los vasodilatadores con capacidad de inhibir la plaqueta (PGI2, NO)165.
El metabolismo transcelular también puede explicar fenómenos clínico asociados a enfermedades trombóticas. Las plaquetas tratadas con aspirina pueden recuperar su capacidad para inducir vasoconstricción, porque pueden metabolizar el LTA2 liberado por neutrófilos a LTC4. El LTC4 es un potente vasoconstrictor y es capaz de sustituir el efecto vasoespásmico del TXA2 en el vaso sanguíneo, propiedad abolida por la aspirina107.
La trombosis tiene un importante componente inflamatorio y ambos están bioquímicamente ligados como parte de un mecanismo general de la defensa del hospedador. Estos procesos, como hemos mencionado, envuelven complejas interacciones multicelulares. El comportamiento físico y biológico de una célula está fuertemente alterado por la proximidad de otras. El resultado final (fenómeno trombótico o antitrombótico) es dictado por las consecuencias del contacto directo de las células, por los productos de secreción de las células implicadas, y el estado de activación de uno o más tipos celulares107.
La activación plaquetaria inducida por los PMN a través de la catepsina G, parece ser el mecanismo por el cual éstos contribuyen al desarrollo de los eventos trombóticos164.
La reactividad plaquetaria in vivo está influida por diferentes factores, particularmente los elevados niveles de lipoproteínas de baja densidad (LDL) y de catecolaminas, los cuales parecen desempeñar un papel en la lesión vascular mediada por las plaquetas155,169.
3.2.3 Metástasis tumorales
La formación de metástasis es un proceso complejo que puede ser influído por interacciones con el sistema hemostático, concretamente con las plaquetas.
La implicación de las plaquetas en el proceso metastático fue inicialmente sugerida por estudios en que se observó que las células tumorales inducían agregación plaquetaria. Las células tumorales pueden causar agregación de las plaquetas, expresando actividad coagulante y generando trombina o liberando ADP170.
Las plaquetas activadas liberan componentes del contenido de sus gránulos tales como péptidos con actividad mitogénica y quimiotáctica y enzimas proteolíticas. Los factores mitogénicos más importantes son el factor de crecimiento derivado de la plaqueta (PDGF) y el factor de crecimiento ß. Ambos péptidos promueven la migración y proliferación de células normales, tales como los leucocitos y fibroblastos, liberación de enzimas proteolíticas y secreción de colágeno. El papel de los factores de crecimiento derivados de la plaqueta es modular la proliferación de la célula tumoral171.
Las proteasas y endoglicosidasas liberadas por las células tumorales, leucocitos y plaquetas son importantes moduladores de la matriz extracelular alrededor del tumor y pueden participar en el proceso de invasión del tumor. Las endoglicosidasas y el PF4 plaquetarios, parecen contribuir a este proceso. Adicionalmente, la trombospondina, una proteína adhesiva liberada principalmente por las plaquetas, ha sido implicada en los procesos de adhesión y puede desempeñar un papel en la invasión y metastización del tumor171.
La adhesión de las células tumorales a la matriz subendotelial, puede ser estimulada por la interacción con las plaquetas activadas, tanto por la liberación de trombina como a través de mecanismos de adhesión mediados por receptores. A la adhesión de las células tumorales deberá seguirle la retracción de las células endoteliales y la adhesión de las células tumorales a la membrana basal subyacente. El tropismo selectivo de la diseminación de las células tumorales puede ser debido a factores de crecimiento específicos o quimiotácticos liberados por los tejidos diana. Un complejo sistema de mediadores derivados de las plaquetas, leucocitos y células tumorales modulan la adhesión de algunas células al endotelio o a la matriz subendotelial. Particularmente relevante parece ser la IL-1 derivada de los leucocitos. Por otra parte, los productos del metabolismo del ácido araquidónico, tanto por la vía de la cicloxigenasa como por la vía de la lipoxigenasa, también desempeñan un papel importante. Por ejemplo, el producto de la vía de la lipoxigenasa 12-HETE estimula la adhesión de las células tumorales a células endoteliales171.
Datos recientes sobre los mecanismos de interacción célula-célula y célula matriz, indican que las plaquetas y las células tumorales poseen moléculas de adhesión similares.
Diversas proteínas adhesivas contienen la secuencia RGD que es reconocida por los receptores de adhesión de la familia de las integrinas. Estos receptores también están presentes en la superficie de las células tumorales y participan en el proceso de motilidad y adhesión, acelerando así la invasión y diseminación tumorales. Las plaquetas poseen diversas integrinas comunes a las células tumorales y endoteliales, implicadas en el proceso de diseminación del tumor. Entre ellas se incluyen la integrina αIIbß3 y el receptor de la vitronectina ya identificadas en diversas líneas de células tumorales172.
Las proteínas adhesivas, fibronectina, vWF, laminina, vitronectina y trombospondina, y los receptores de integrinas están implicados en la adhesión tumoral, en las interacciones plaqueta-tumor, y en las metástasis. Las plaquetas se adhieren a las células tumorales a través de la integrina αIIßb3 vía puentes de fibronectina y vWF172.
Las plaquetas contribuyen probablemente a las metástasis deteniendo las células tumorales en la vasculatura, a través de la formación de trombos plaqueta-tumor y/o adhesión de plaquetas-tumor a la matriz subendotelial172.
Las plaquetas activadas protegen las células tumorales adheridas de las células Natural Killer y reducen los efectos citotóxico del TNF α en las células tumorales173,174. Por otra parte, se propuso la hipótesis de que los trombos plaqueta-tumor pueden contribuir a la fijación del tumor, creando necrosis subendotelial distal con consiguiente exposición de la matriz subendotelial, sustrato más eficiente para la adhesión del tumor172. Las plaquetas también pueden contribuir a la metástasis generando trombina en su superficie. Estudios recientes indican que el receptor de la trombina está presente en diversas líneas de células tumorales y cuando está ocupado y proteolizado, lleva al incremento de la adhesividad del tumor a las plaquetas, vWF y fibronectina, así como incrementa las metástasis in vivo de 10-156 veces172.
La mayor adhesividad tras la estimulación con trombina, puede ocurrir vía GPIIbIIIa presente en la célula tumoral. Además, la trombina puede activar las metástasis actuando como un agente mitogénico o induciendo la formación de uniones de tipo gap, que facilitan la adhesión de las células tumorales a la matriz subendotelial más adherente172.
Los datos disponibles indican que es posible que la generación de trombina en bajo grado puede ser peligrosa para algunos pacientes con cáncer, ya que predispone a la progresión metastática de la lesión. Por otra parte, hay evidencias de que muchos tumores pueden activar la coagulación con generación de trombina, produciendo el potente coagulante factor tisular175.
Hace mucho que se utilizan agentes antiplaquetarios en el tratamiento de las metástasis tumorales. El efecto antimetastático de la aspirina fue observado por algunos investigadores, pero no confirmado por otros. La ticlopidina, otro potente inhibidor de la activación plaquetaria, fue incapaz de inhibir las metástasis en modelos experimentales. Más eficaces parecen ser los análogos de la pGI2 y los inhibidores de la tromboxano sintetasa. Además, han sido utilizados con éxito los bloqueadores de canales de Ca2+ y, más recientemente, los antagonistas de GPIIbIIIa. Los anticuerpos monoclonales contra GPIIbIIIa inhiben la adhesión de las células tumorales y la agregación plaquetaria inducida por las células tumorales171.
Bibliografía
1. Crawford N, Scrutton MC. Biochemistry of blood platelet. In: AL Bloom, DP Thomas (Editors). Haemostasis and thrombosis. NY, Churchill Livingstone; 1987. p. 47-77. [ Links ]
2. Bennett JS, Kolodziej MA. Disorders of platelet function. Dis Mon 1992; 38 (8): 577-631. [ Links ]
3. Nesheim ME, Furmaniak-Kazmerczak E, Henin C, Côte G. On the existence of platelet receptors for factor V(a) and factor VIII(a). Thromb Haemost 1993; 70: 80-86. [ Links ]
4. Winthrobe MM. Blood, pure and eloquen. NY, McGraw Hill, 1980. [ Links ]
5. Coller BS. Disorders of platelets.In: OD Ratnoff, CD Forbes (editors). Disorders of hemostasis. Orlando, Grune & Stratton; 1984. p. 73-176. [ Links ]
6. Moriau M, Lavenne EP, Scheiff JM, Debeys CC. The relationship between endothelium, subendothelium, and platelets. In: M Moriau, EP Lavenne, JM Scheiff, CC Debeys (editors). Blood platelets. Paris, Halograme, 1990. p. 13-49. [ Links ]
7. Nurden AT. Platelet membrane glycoproteins and their clinical aspects. In: M Verstraete, J Vermylen, R Lijnen, J Arnout (editors). Thrombosis and Haemostasis. Leuven, Int Soc Haem Thromb and Leuven University Press, 1987. p. 93-125. [ Links ]
8. Nieuwenhuis HK, Oosterhout JJG, Rozemuller E, Iwaarden F, Sixma JJ. Studies with a monoclonal antibody aginst activated platelets: evidence that a secreted 53.000-molecular weight lysosome-like granule protein is exposed on the surface of activated platelets in the circulation. Blood 1987; 70 (3): 838-845. [ Links ]
9. Stenberg PA, McEver R, Shuman MA, Jaques YV, Baiton DF. A plate let alpha-granule membrane protein (GMP-140) isexpressed on the plasma membrane after activation. J Cell Biol 1985; 101: 880-886. [ Links ]
10. Kieffer N, Phillips DR. Platelet membrane glycoproteins: function in cellular interactions. Ann Rev Cell Biol 1990; 6: 329-357. [ Links ]
11. Bennett JS. Molecular biology of platelet membrana glycoproteins. Blood 1990; 27: 186-204. [ Links ]
12. Peerschke EIB. Platelet membrane glycoproteins. Functional characterization and clinical applications. Clin Pathol 1992; 98 (4): 455-463. [ Links ]
13. Schmitz G, Rothe G, Ruf A, Barlage S, Tschope D, Clemetson KJ, Goodall AH, Michelson AD, Nurden AT, Shankey TV. European working group on clinical cell analysis: Consensus protocol for the flow cytometric characterisation of platelet function. Thromb Haemost 1998; 79: 885-896. [ Links ]
14. Lam SCT, Plow F, Ginsberg MH. Platelet membrane glycoprotein IIb heavy chain forms a complex with glycoprotein IIIa that binds Arg-Gly-Asp peptides. Blood 1989; 73 (6): 1513-1518. [ Links ]
15. Ruoslahti E. Integrins.J Clin Invest 1991; 87: 1-5. [ Links ]
16. Ginsberg MH, Xioping D, O'Toole TE, Loftus JC, Plow EF. Platelet integrins. Thromb Haemost 1993; 70 (1): 87-93. [ Links ]
17. Shattil SJ, Hoxie JA, Cunningham M, Brass LF. Changes in the platelet membrane glycoprotein iib. Iiib complex during platelet activaion. J Biol Chem 1985; 260 (20): 11107-11114. [ Links ]
18. Kunicki TJ, Pidard D, Jean-Philippe Rosa, Nurden AT. The formation of Ca++ - dependent complexes of platelet membrane glycoproteins IIb and IIIa in solution as determined by crossed immunoelectrophoresis. Blood 1981; 58 (2): 268-278. [ Links ]
19. Phillips DR, Charo IF, Scarborough RM. GPIIb-IIIa: the responsive integrin. Cell 1991; 65: 359-362. [ Links ]
20. Shattil SJ. Regulation of platelet anchorage and signaling by integrin aIIb b3. Thromb Haemost 1993; 70 (1): 224-228. [ Links ]
21. Parise LV, Criss AB, Nannizi L, Wardell M. Glycoprotein IIIa is phosphorylated in intact human platelets.Blood 1990; 75 (12): 2363- 2368. [ Links ]
22. Fujimoto T, Fujimura K, Kuramoto A. Functional Ca2+channel produced by purified platelet membrane glycoprotein IIb-IIIa complex incorporated in to planar phospholipid bilayer. Thromb Haemost 1991; 66 (5): 598-603. [ Links ]
23. Bachelot C, Cano E, Grelac F, Saleun S, Druker BJ, Levy-Toledano S, Fischer S, Rendu F. Functional implications of tyrosine protein phosphorylation in platelets. Simultaneous studies with different agonists and inhibitors. Biochem J 1992; 284 (3): 923-928. [ Links ]
24. Jennings LK, Ashmun RA, Wang WC, Dockter ME. Analysis of human platelet glycoproteins IIb-IIIa and glanzmann's trombasthenia in whole blood by cytometry. Blood 1986; 68 (1): 173-179. [ Links ]
25. Hagen I, Nurden A, Bjerrum OJ, Solum NO, Caen J. Immunochemical evidence for protein abnormalities from platelets with Glanzmann's thrombasthenia and Bernard-Soulier Syndrome. J Clin Invest 1979; 65: 722-731. [ Links ]
26. Kato A, Yamamoto K, Miyazaki S, Jung SM, Moroi M, Aoki N. Molecular basic for glanzmann's thrombasthenia (GT) in a compound heterozygote with glycoprotein IIb gene: a proposal for the classification of GT based on the biosynthetic pathway of glycoprotein IIb-IIIa complex. Blood 1992; 79 (12): 3212-3218. [ Links ]
27. Clemetson KJ. Platelets receptors and funcional disorders. Schweiz Med Wschr 1991; 121 (Suppl 43): 12-23. [ Links ]
28. Catimel B, Parmentier S, Leung LL, McGregor JL. Separation of important new glycoproteins (GPIa, GPIc, GPIc*, GPIIa and GMP140) by f.p.l.c. Characterization by monoclonal antibodies and gas-phase sequencing. Biochem J 1991; 279 (2): 419-425. [ Links ]
29. Preissner KT, Jenne D. Structure of vitronectin and its biological role in haemostasis. Thromb Haemost 1991; 66 (1): 123-132. [ Links ]
30. Modderman PW, Admiraal LG, Sonnenberg A, von-dem-Borne AE. Glycoproteins V and Ib-IX form a noncovalent complex in the platelet membrane. J Biol Chem 1992; 267 : 364-369. [ Links ]
31. Roth GJ. Developing relationships: arterial platelet adhesion, glycoprotein Ib, and leucine-rich glycoproteins. Blood 1991; 77 (1): 5-19. [ Links ]
32. Meyer D, Girma JP. Von Willebrand factor: structure and function. Thromb Haemost 1993; 70 (1): 99-104. [ Links ]
33. Fressinaud E, Meyer D. von Willebrand factor and platelet interactions with the vessel wall. Blood Coag Fibrinolysis 1991; 2: 333-340. [ Links ]
34. Ruggeri ZM. The platelet glycoprotein Ib-IX complex. Prog Hemost Thromb 1991; 10: 35-68. [ Links ]
35. Fox JEB, Reynolds CC, Johnson MM. Identification of glycoprotein Ib as one of the major proteins phosphorylated during exposure of intact agents that that activate cyclic AMP-dependent kinase. J Biol Chem 1987; 262: 12627-12631. [ Links ]
36. Michelson AD. Thrombin-induced down-regulation of the platelet membrane glycoprotein Ib-IX complex. Sem Thromb Haemost 1992; 18 (1): 18-27. [ Links ]
37. Nurden P. Bidirectional trafficking of membrane glycoproteins following platelet activation in suspension. Thromb Haemost 1997; 78: 1305-1315. [ Links ]
38. Terninisien C, Jandrot-Perrus M, Huisse MG, Guillin MC. Effect of phosphopyridoxylation on thrombin interaction with platelet glycoprotein Ib. Blood Coag Fibrinolysis 1991; 2: 521-528. [ Links ]
39. Nurden AT, Dupuis D, Kunicki TJ, Caen JP. Analysis of the glycoprotein and protein composition of Bernard-Soulier platelets by single and two-dimensional sodium dodecyl sulfate-polyacrylamide gel electrophoresis. J Clin Invest 1981; 67: 1431-1440. [ Links ]
40. Clemetson KJ, McGregor JL, James E, Dechavanne M, Lüscher EF. Characterization of platelet membrane glycoprotein abnormalities in Bernard-Soulier Syndrome and comparation with normal by surface-labeling techniques and high-resolution two-dimensional gel electrophoresis. J Clin Invest 1982; 70: 304-311. [ Links ]
41. George JN, Picket EB, Saucerman S, McEver RP, KUnicki TJ, Kieffer N, Newman PJ. Platelet surface glycoproteins. Studies on resting and activated platelets in patients during adult respiratory distress syndrom and cardiac surgery. J Clin Invest 1986; 78: 340-348. [ Links ]
42. Berger G, Caen JP, Berndt MC, Cramer EM. Ultrastructural demonstration of CD36 in the a-granule membrane of human platelets and megakaryocytes. Blood 1993; 82 (10): 3034-3044. [ Links ]
43. Enouf J, Bobe R, Lacabaratz-Porret C, Bredoux R, Corvazier E, Kovàcs T, Papp B. The platelet Ca2+ tranport ATPase system. Platelets 1997; 8: 5-13. [ Links ]
44. Salzman EW, Ware JA. Ionized calcium as an intracellular messenger in blood platelets. Prog Hemost Thromb 1989; 9: 177-202. [ Links ]
45. Dean WL, Sullivan DM. Structural and functional properties of a Ca2+-ATPase from human platelets. J Biol Chem 1982; 257 (23): 14390- 14394. [ Links ]
46. Furman MI, Gardner TM,Goldschmidt-Clermont PJ. Mechanisms of cytoskeletal reorganization during platelet activation. Thromb Haemost. 1993; 70 (1): 229-232. [ Links ]
47. Lind SE. Platelet morphology. In: AL Bloom, CD Forbes, DP Thomas, EGD Tuddenhan (editors.) Haemostasis and thrombosis. NY , Churchill Livingstone, 1994. p. 201-218. [ Links ]
48. Tunnacliffe A, Majumdar S, Yan B, Poncz M. Genes for ß-thromboglobulin and platelet factor 4 are closely linked and form part of a cluster of related genes on chromosome 4. Blood 1992; 79 (11): 2896-2900. [ Links ]
49. BretonGorius J, Clezardin P, Guichard J et al. Localization of platelet osteonectin at the internal face of alfa-granule membranes in platelets and megacaryocytes. Blood 1992; 79: 936-941. [ Links ]
50. Bithell TC. The physiology of primary hemostasis. In: GR Lee, TC Bithell, J Foerster, JW Athens, JN Lukens (editors). Wintrobe's Clinical Hematology. Londres, Lea & Febiger, 1993. p. 511-616. [ Links ]
51. Corash L. The relationship between magakaryocyte ploidy and platelet volume. Blood Cells 1989; 15: 81-107. [ Links ]
52. Handagama PJ, Shuman MA, Bainton DF. The origin of platelet granule proteins. Prog Clin Biol Res 1990; 356: 119-130. [ Links ]
53. Schick PK, Wojenski CM, Bennett VD, Ivanova T. The synthesis and localization of alternatively spliced fibronection EIIIB in Resting and thrombin-treated megakaryocytes. Blood 1996; 87 (5): 1877-1823. [ Links ]
54. Norol F, Vitrat N, Cramer E, Guichard J, Burstein AS, Vainchenker W, Debili N. Effects of cytokines on platelet production from blood and marrow CD34+ cell. Blood 1998; 91(3): 830-843. [ Links ]
55. Penington DG. Formation of platelets. Cell Tissue Physiol 1981; 5: 19-41. [ Links ]
56. Stanley ER, Jubinsky PT. Factors affecting the growth and differentiation of haematopoietic cells in culture. Clin Haematol 1984; 13 (2): 329-348. [ Links ]
57. Trowbridge EA. Pulmonary platelet production : a physical analog of mitosis. Blood Cells 1988; 13: 451-465. [ Links ]
58. Metcalf D. Trombopoietina - at last. Nature 1994; 369: 519-520. [ Links ]
59. Wendling F, Maraskovsky E, Debili N, Florindo C, Teepe M, Titeux M, Methia N, Breton-Gorius J, Cosman D, Vainchenker W. c-Mpl ligand is a humoral regulator of megakaryocytopoiesis. Nature 1994; 369: 571-574. [ Links ]
60. Nichol JL, Hokom AC, Hornkol A et al. Megakaryocyte growth and development factor. Analysis of in vitro effects of human megakaryopoiesis and endogenous serum levels during chemotherapy induced thrombocytopenia. J Clin Invest 1995; 95: 2973-2978. [ Links ]
61. Xi X, Caen JP, Fournier S. Direct and reversible inhibition of platelet factor 4 on megakaryocyte development from CD34+ cord blood cells: comparative studies with transforming growth factor ß1. Br J Haematol 1996; 93: 265-272. [ Links ]
62. Corash L, Tan H, Gralnick HR. Heterogeneity of human whole blood platelet subpopulations. I. Relationship between buoyant density, cells volume, and ultrastructure. Blood 1977; 49 (1): 71-87. [ Links ]
63. Bessman JD. Platelet turnover. In: Haemostasis and thrombosis. AL Bloom, CD Forbes, DP Thomas, EGD Tuddenhan (eds.). Churchill Livingstone, NY 1994; pp 195-200. [ Links ]
64. Siess W. Molecular mechanisms of platelet activation. Physiol Rev 1989; 69 (1): 58-178. [ Links ]
65. Haslam RJ. Signal tansduction in platelet activation. In: M Verstraete, J Vermylen, R Lijnen, J Arnout (editors). Thrombosis and Haemostasis. Leuven, Int Soc Haem thromb and Leuven University Press, 1987. p.147-174. [ Links ]
66. Brass LF, Hoxie JA, Maning DR. Signaling through G proteins and G protein-coupled receptors during platelet activation. Thromb Haemost 1993; 70 (1): 217-223. [ Links ]
67. Brass FL, Manning DR, Shatil JS. GTP-Binding proteins and platelet activation. Progr Thromb Hemost 1991; 10: 127-174. [ Links ]
68. Brass LF, Manning DR, Cichowski K, Arams CS. Signaling throught G proteins in platelets: to the integrins and beyond. Thromb Haemost 1997; 78 (1): 581-589. [ Links ]
69. Akkerman JWN, van Willigen G. Platelet activation via trimeric GTP-binding proteins. Haemostasis. 1996; 26 (4): 199-209. [ Links ]
70. Aktories K, Jackogs KH. Ni-mediated inhibition of human adenylcyclase by thrombin. Eur J Biocherm 1984; 145: 333-338. [ Links ]
71. Brass LF, Shaller CC, Belmonte EJ.Inositol 1,4,5-triphosphate-induced granule secretion in platelets. Evidence that the activation of phospholipase C mediated by thromboxane receptores involves a guanine nucleotide binding protein-dependent mechanism distinct from thrombin. J Clin Invest 1987; 79: 1269-1275. [ Links ]
72. Berridge MJ. Inositol triphosphate and Ca2+ signaling. Nature 1993; 361: 315-325. [ Links ]
73. Brass LF, Joseph SK. A role inositol triphosphate in tntracellular Ca2+ mobilization and granule secretion in platelets. J Biol Chem 1985; 260 (28): 15172-15179. [ Links ]
74. Adunyah S, Dean W. Inositol triphosphate induce Ca2+ release from human membranes. Biochem Biophys Res Comun 1985; 128: 1274-1280. [ Links ]
75. Authi KS, Crawford N. Inositol 1,4,5- trisphosphate-induced realease of sequestered Ca2+ from highly purified human platelet intracellular membranes. Biochem J 1985; 230: 247-253. [ Links ]
76. O´Rourke F, Zavoico GB, Smith LH, Feinstein MB. Stimulus-response coupling in a cell-free platelet system GTP-dependent release of Ca2+ by thrombin and inhibiton with a monoclonal antibody that blocks calcium release by IP3. FEBS Lett 1987; 214: 176-180. [ Links ]
77. Daniel JL, Molish IR, Rigmaiden M, Stewart G. Evidence for role of myosin phosphorylation in the initiation of the platelet shape change response. J Biol Chem 1984; 259: 9826-9831. [ Links ]
78. Adelstein RS. Regulation of contractile proteins by phosphorylation. J Clin Invest 1983; 72: 1863-1866. [ Links ]
79. Broekman MJ, Ward JW, Marcus AJ. Phospholipid metabolism in stimulated human platelets. Changes in phosphatidylinositol, phosphatidic acid, and lysophospholipids. J Clin Invest 1980; 66: 275-283. [ Links ]
80. Rana RS, Hokin LE. Role of phosphoinositides in transmembrane signaling. Physiol Rev 1990; 70 (1): 115-164. [ Links ]
81. Kaibuchi K, Sawamura YM, Hoshijima M, Fujikura, Nishizuka Y. Synergistic functions of protein phosphorylation and calcium mobilization in platelet activation. J Biol Chem 1983; 258: 6701-6704. [ Links ]
82. Nishizuka Y. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature 1984; 308: 693-698. [ Links ]
83. Shattil S, Brass L. Induction of the fibrinogen receptor on human platelets by intracellular mediators. J Biol Chem 1987; 262: 992-1000. [ Links ]
84. Benton AM, Gerrard JM, Michiel T, Kindom SE. Are lysophosphatidic acids involved in stimulus activation coupling platelets? Blood 1982; 60 (3): 642-649. [ Links ]
85. Pollock WK, Armstrong RA, Brydon LJ, Jones RL, MacIntyre DE. Thromboxane -induced phosphatidate formation in hunan platelets. Relation to receptor occupancy and to changes in cytosolic free calcium. Biochem J 1984; 219: 833-842. [ Links ]
86. Fox JEB. Platelet activation: new aspects. Haemostasis 1996; 26 (suppl 4): 120-131. [ Links ]
87. Shattil SJ, Kashiwagi H, Pampori N. Integrin signaling: The platelet paradigm. Blood 1998; 91 (8): 2645-2657. [ Links ]
88. Hofmann SL, Majerus PW. Modulation of phosphatidylinositol-specific phospholipase C activity by phospholipid interactions, diglycerides, and calcium ions. J Biol Chem 1982; 257 (23): 14359-14364. [ Links ]
89. Bansal VS, Inhorn RC, Majerus PW. The metabolism of inositol 1,3,4-triphosphate to inisitol 1,3-biphosfate. J Biol Chem 1987; 262 (25): 9444-94447. [ Links ]
90. Daniel JL, Dangelmaier CA, Smith JB. Formation and metabolism of inositol 1,4,5-triphosphate in human platelets. Biochem J. 1987; 246: 109-114. [ Links ]
91. Fox JEB. The platelet cytoskeleton. Thromb Haemost 1993; 70: 884-893. [ Links ]
92. Bruin T, Asijee GM, Prins A, ten Cate JW. Subcellular distribution and phosphorylation of vinculin isoforms in human blood platelets. Thromb Haemost 1991; 65 (2): 206-211. [ Links ]
93. Lehninger AL, Nelson DL, Cox MC. Principles of Biochemistry. NY, Worth Publishers, 1993. [ Links ]
94. Naka M, Nishikawa M, Adelstein RS, Hidaka H. Phorbol ester-induced activation of human platelets is associated with protein kinase C phosphorylation of myosin light chains. Nature 1983; 306: 490-492. [ Links ]
95. Brass LF, Woolkais MJ, Manning DR. Interactions in platelets between G proteins and agonists that stimulate phospholipase C and inhibit adenylciclase. J Biol Chem 1988; 263: 5348-5355. [ Links ]
96. Connoly TM, Bansal VS, Bross TE, Irvine Rf, Majerus PW. The metabolism of tris and tetraphosphates of inositol by 5- phosphomonoesterase and 3-cinase enzimes. J Biol Chem 1987; 262 (5): 2146-2149. [ Links ]
97. Kroll MH. Mechanisms of platelet activation. In: AL Bloom, CD Forbes, DP Thomas, EGD Tuddenhan. Haemostasis and thrombosis (editors). NY, Churchill Livingstone, 1994. p. 247-277. [ Links ]
98. Bell RM, Burns DJ. Lipid activation of protein kinase. J Biol Chem 1991; 266:4661-4664. [ Links ]
99. Brace LD, Vention DL, Breton BC. Thromboxane A2 prostaglandin H2 mobilizes calcium in human blood platelets. Circulation 1982; Suppl 64: 1198a. [ Links ]
100. Mayer RJ, Marshall LA. New insights on mammalian phospholipase A2(s); comparison of arachidonoyl-selective and-nonselective enzimes. FASEB 1993; 7: 339-348. [ Links ]
101. Sweatt JD, Connolly TM, Cragoe EJ, Limbird LE. Evidence that Na+/H+ exchange regulates receptor-mediated phospholipase A2 activation in human platelets. J Biol Chem 1986; 26: 8667-8673. [ Links ]
102. Levy-Toledano S, Gallet C, Nadal F, bryckaert M, Maclouf J, Rose J-F. Phosphorylation and dephosphorylation mechanisms in platelet function: a tightly regulated balance. Thromb Haemost 1997; 78 (1): 226-233. [ Links ]
103. Ferrell JE, Martin GS. Tyrosine-specific protein phosphorylation is regulated by thrombin. Mol Cell Biol 1988; 8: 3603-3610. [ Links ]
104. Golden A, Brugge JS. Thrombin treatment induces rapid changes in tyrosine phosphorylation in platelets. Proc Natl Acad Sci USA 1989; 86: 901-905. [ Links ]
105. Tao J, Johansson JS, Hayenes DH. Stimulation of dense tubular Ca2+ uptake in human platelets by cAMP. Biochem Biophys Acta 1992; 1105 (1): 29-39. [ Links ]
106. Friebe A, Schultz G, Koesling D. Stimulation of soluble guanylate cyclase by superoxide dismutase is mediated by NO. Biochem J 1998; 335: 527-531. [ Links ]
107. Marcus AJ, Safier LB, Broekman JB, Islam N, Fliesssbach JH,Hajjar CA, Kaminski WE, Jendraschak E, Silverstein RL, Schacky C. Thrombosis and inflammation as multicellular processes: significance of cell-cell interactions. Thromb Haemost 1995; 74 (1): 213-217. [ Links ]
108. Adunyah SE, Dean WL. Ca2+ transport in human platelet membranes. J Clin Invest 1986; 261: 3122-3127. [ Links ]
109. Sargeant P, Sage SO. Calcium signalling in platelets and other nom-excitable cells. Pharmacol Therap 1994; 64: 395-443. [ Links ]
110. Sage SO, Merritt JE, Mahaut-Smith MP, Rink TJ. Agonist-evoked Ca2+ entry in human platelets. Biochem J 1992; 285: 341-343. [ Links ]
111. Berridge MJ. Review article capacitative calcium entry. Biochem J 1995; 312: 1-11. [ Links ]
112. Gear ARL, Schneider W. Control of platelet glycogenolysis. Activation of phosphorylase kinase by calcium. Bichim Biophys Acta 1975; 392: 111-120. [ Links ]
113. Courcelles DC, Roevens P, Verheyen F, Belle HV, De Clerck F. The effect of the intracellular calcium chelator quin-2 on platelet phosphoinositide metabolism, protein phosphorylation and morphology. Thromb Haemost 1987; 58 (3): 927-931. [ Links ]
114. Knight DE, Hallam TJ, Scrutton MC. Agonist selectivity and second messenger concentration in Ca2+ -mediated secretion. Nature 1982; 296: 256-257. [ Links ]
115. Fressinaud E, Baruch D, Rothschild C, Baumgartner HR, Meyer D. Platelet von Willebrand factor: evidence for its involvement in platelet adhesion to collagen. Blood 1987; 70: 1214-1217. [ Links ]
116. Ruggeri ZM, De-Marco L, Gatti L, Bader R, Montgomery RR. Platelets have more than one binding site for von Willebrand factor. J Clin Invest 1983; 72 (1): 1-12. [ Links ]
117. Kehrel B. Platelet - collagen interactions. Semin Thromb Haemost 1995; 21 (2): 123-129. [ Links ]
118. White JG, Rao GHR. Microtubule coils versus the surface membrane cytoskeleton in maintenance and restoration of platelet discoid shape. Am J Pathol 1998; 152 (2): 597-609. [ Links ]
119. Winokur R, Hartwig JH. Mechanism of shape change in chilled human platelets. Blood 1995; 85 (7): 1796-1804. [ Links ]
120. Harrison P, Savidge GF, Cramer EM. The Origin and Physiological Relevance of a-Granule Adhesive Proteins. Brit J Haematol 1990; 74: 125-130. [ Links ]
121. Hourani SMO, Cusack NM. Pharmacological Receptors on blood platelets. Pharmacol Rev 1991; 43 (3): 243-298. [ Links ]
122. Lawrence AH, Kenneth GM. Thrombosis and fibrinolysis. In: Thrombosis and cardiovascular disorders. V Fuster e M Verstaete (eds). WB Sauders Company 1986; pp 1-16. [ Links ]
123. Peerschke EIB. Platelet membranes and receptores. In: Haemostasis and thrombosis. AL Bloom, CD Forbes, DP Thomas, EGD Tuddenhan (eds.). Churchill Livingstone, NY 1994; pp 219-245. [ Links ]
124. Metzelaar MJ, Clevers HC. Lysossomal membrane glycoproteins in platelet. Thromb Haemost 1992; 68 (4): 378-382. [ Links ]
125. Israels SJ, Gerrard JM, Jacques YV, McNicol A, Cham B, Nishibori M, Baiton DF. Platelet dense granule membranes contain both granulophysin and P-selectin (GMP-140). Blood 1992; 80 (1): 143-152. [ Links ]
126. Gerrard JM, Lint D, Sims PJ, Wiedmer T, Fugate RD, McMillan E, Bobertson C, Israels SJ. Identification of a platelet dense granule membrane protein that is deficient in a patient with the Hermansky-Pudlak syndrome. Blood 1991; 77 (1): 101-112. [ Links ]
127. Metzelaar MJ, Heijnen HFG, Sixma JJ, Nieuwenhuis HK. Identification of a 33-Kd protein associated with the a-granule membrane (GMP-33. that is expressed on the surface of activated platelets. Blood 1992; 79 (2): 372-379. [ Links ]
128. Bruijne-Admiraal LG, Modderman PW, von der Borne AEGK, Sonnenberg A. P-selectin mediates Ca2+- dependent adhesion of activated platelets to many different types of leukocytes: detection by flow cytometry. Blood 1992; 80 (1): 134-142. [ Links ]
129. McEver RP. Properties of GMP-140, an inducible granule membrane protein of platelets and endothelium. Blood Cells 1990; 16: 73-83. [ Links ]
130. Wagner DD. The Weibel-palade body: the storage granule for von Willebrand factor and P - selectin. Thromb Haemost 1993; 70 (1): 105-110. [ Links ]
131. Hamburger SA, McEver RP. GMP-140 mediates adhesion of stimulated platelets to neutrophils. Blood 1990; 75 (3): 550-554. [ Links ]
132. Metzelaar MJ, Wijngaard PLJ, Peters PJ, Sixma JJ, Nieuwenhuis HK, Clevers HC. CD63 antigen. A novel Lysosomal membrane glycoprotein cloned by a screening procedure for intracellular antigens in eukaryotic cells. J Biol Chem 1991; 266 (5): 3239-3245. [ Links ]
133. Krishnasvamy S, Mann KG. The binding of factor Va to phospholipid vesicles. J Biol Chem 1988; 263: 5714-5723. [ Links ]
134. Bevers EM, Comfurius P, Van Rijin JLML. Generation of prothrombin converting activity and the exposure of phosphatidyl-serine at the outer surface of platelets. Eur J Biochem 1982; 122: 429-436. [ Links ]
135. Hoffman M, Monroe D M, Roberts HR. Procoagulant assembly on platelets in a tissue factor initiated system. Brit J Haematol 1994; 87: 203a. [ Links ]
136. Walsh PN, Baglia FA, Jameson BA. Factor XI and platelets: activation and regulation. Thromb Haemost 1993; 70 (1): 75-79. [ Links ]
137. Kenneth A. Flow cytometric measurement of platelet-associated immunoglobulin. Pathol Immun Res 1987; 7: 395-408. [ Links ]
138. Zwaal RFA, Schroit AJ. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood 1997; 89 (4): 1121-1132. [ Links ]
139. Dachary-Prigent J, Pasquet J-M, Freyssinet J-M, Nurden AT. Calcium involvement in aminophosphoslipid exposure and microparticle formation during platelet activation: a study using Ca2+- ATPase inhibitors. Biochemistry 1995; 34: 11625-11634. [ Links ]
140. Pasquet J-M, Dachary-Prigent J, Nurden AT. Calcium influx is a determining factor of calpain activation and microparticle formation in platelets. Eur J Biochem 1996; 239: 647-654. [ Links ]
141. Cristalli G, Mills DC. Identification of a receptor for ADP on blood platelets by photoaffinity labelling. Biochem J 1993; 291 (pt 3): 875-881. [ Links ]
142. Coughlin SR. Thrombin receptor structure and function. Thromb Haemost 1993; 66 (1): 184-187. [ Links ]
143. Grand RJA, Turnell AS, Grabham PW. Cellular consequences of thrombin-receptor activation. Biochem J 1996; 313: 353-368. [ Links ]
144. McNicol A, Gerrard JM. Post - receptor events associated with thrombin -induced platelet activation. Blood Coag Fibrinolysis 1993; 4: 975-991. [ Links ]
145. Clemetson KJ. Platelet activation: Signal transduction via membrane receptors. Thromb Haemost 1995; 74 (1): 111-116. [ Links ]
146. Puri RN, Colman RF, Colman RW. Platelet activation by 2 - (4-bromo-2,3-Dioxobutylthiol. adenosine 5' -diphosphate is mediated by its binding to a putative ADP receptor, aggregin. Eur J Biochem 1996; 236: 862-870. [ Links ]
147. Kunapuli SP, Daniel JL. P2 receptor subtypes in the cardiovascular system. Biochem J 1998; 336: 513-523. [ Links ]
148. Gachet C, Hechler B, Leon C, Vizi C, Lepay C, Ohlman P, Cazenave J. Activation of ADP receptors and platelet function. Thromb Haemost 1997; 78 (1): 271-275. [ Links ]
149. Jantzen H, Gousset L, Bhaskara V, Vicent D, Tai A, Reynolds EE, Conley PB. Evidence for two distinct G-protein-coupled ADP receptors mediating platelet activation. Throm Haemost 1999; 81: 111-117. [ Links ]
150. Hechler B, Léon C, Vial C, Vigne P, Frelin C, Cazenave J-P, Gachet C. The P2Y1 receptor is necessary for adenosine 5'-diphosphate-induced platelet agrregation. Blood 1998; 92 (1): 152-159. [ Links ]
151. Jin J, Kunapuli P. Coactivation of two different G protein-coupled receptors is essential for ADP-induced platelet aggregation. Cell Biol 1998; 95: 8070-8074. [ Links ]
152. Gachet C, Cazenave JP, Ohlman P, Bouloux C, Defreyn G, Driot F, Maffrand JP. The thienopyridine ticlopidine selectively prevents the inhibitory effects of ADP but not adrenaline on AMPc levels raised by stimulation of adenylate cyclase of human platelets by PGE1. Biochem Pharmacol 1990; 40: 2683-2687. [ Links ]
153. Blockmans D, Deckmyn H, Vermylen J. Platelet activation. Blood Reviews 1995; 9: 143-156. [ Links ]
154. Chao W, Olson MS. Platelet-activation factor: receptors and signal transduction. Biochem J. 1993; 292: 617-629. [ Links ]
155. Anfossi G, Trovati M. Role of catecholamines in platelet function: pathophysiological and clinical significance. Eur J Clin Invest. 1996; 26: 353-370. [ Links ]
156. Mann KG, Tracy PB, Jenny RJ, Odegaard BH, Nesheim ME. Platelets and Coagulation.In: Thrombosis and Haemostasis. Verstraete M, Vermylen J, Lijnen HR, Arnout J (Eds). Leuven University Press, Leuven 1987. [ Links ]
157. Pujol-Moix N. In: Trombocitopenia. Mosby, Madrid, 1995; pp 32-40. [ Links ]
158. Sims PJ, Wiedmer T. The response of human platelets to activated components of the complement system. Immunol Today 1991; 12: 338-342. [ Links ]
159. Sims PJ, Faioni EM, Wiedmer T, Shattil SJ. Complement proteins C5b-9 cause release of membrane vesicles from the platelet suface that are enriched in the membrane eceptor for coagulation factor Va and express prothrombinase activity. J Biol Chem 1988; 263 (34. 18205-18212. [ Links ]
160. Gentry PA. The mammalian blood platelet: its role in haemostasis, inflammation and tissue repair. J Comp Path 1992; 107: 243-270. [ Links ]
161. Page CP. Platelets as inflammatory cells. Immunopharmacol 1989; 17: 51-57. [ Links ]
162. Hawrylowicz CM. Viewpoint: a potential role for platelet derived cytokines in the inflammatory response. Platelets 1993; 4: 1-10. [ Links ]
163. Evangelista V, Celardo A, Elba GD, Manarini S, Miranov A, Gaetano G, Cerletti C. Platelet contribution to leukotriene production in inflammation: in vivo evidence in the rabbit. Thromb Haemost 1999; 81: 442-448. [ Links ]
164. Cerletti C, Evangelista V, Molino M, Gaetano G. Platelet activation by polymorphonuclear leukocytes: role of cathepsin G and P-selectin. Thromb Haemost 1995; 74 (1): 218-223. [ Links ]
165 . Wu KK. Platelet activation mechanisms and markers in arterial thrombosis. J Int Med 1996; 239: 17-34. [ Links ]
166. Marcus AJ. Cellular interactions of platelets in thrombosis. In: AL Bloom, CD Forbes, DP Thomas, EGD Tuddenhan (editors.). Haemostasis and thrombosis. NY, Churchill Livingstone; 1994. p. 279-289. [ Links ]
167. Ruggeri ZM. Mechanisms of shear-induced platelet adhesion and aggregation. Thromb Haemost 1993; 70: 119-123. [ Links ]
168. Ruggeri ZM, Dent JA, Saldívar E. Contribution of distinct adhesive interactions to platelet aggregation in flowing blood. Blood 1999; 94 (1): 172-178. [ Links ]
169. Aviram M. LDL-Platelet interaction under oxidative stress induces macrophage foam cell formation. Thromb Haemost 1995; 74 (1): 560-564. [ Links ]
170. Heinmoller E, Weinel RJ, Heidtmann HH, Salge U, Seitz R, Schmitz I, Muller KM, Zirngibl H. Studies on tumor-cell-induced platelet aggregation in human lung cancer cell lines. J Cancer Res Clin Oncol 1996; 122: 735-744. [ Links ]
171. Poggi A, Donatti MB. Platelets and tumor metastasis. In: The platelet in health and disease. Page CP (ed), Blackwell Scientific Publications Oxford 1991; pp 175-190. [ Links ]
172. Nierodzik ML, Klepfish A, Karpatkin S. Role of platelets, thrombin, Integrin IIb-IIIa, fibronectin and Von Willebrand factor on tumor adhesion in vitro and metastasis in vivo. Thromb Haemost 1995; 74 (1. 282-290. [ Links ]
173. Philippe C, Philippe B, Fouqueray B, Perez J, Lebret M, Baud L. Protection from tumor necrosis factor-mediated cytolysis by platelets. Am J Pathol 1993; 143: 1713-1723. [ Links ]
174. Shau H, Roth MD, Golub SH. Regulation of natural killer function by nonlymphoid cells. Nat Immun 1993; 12: 235-249. [ Links ]
175. Biggerstaff JP, Seth NB, Meyer TV, Amirkosravi A, Francis JL. Fibrin monomer increases platelet adherence to tumor cells in a flowing system: a posssible role in metastasis. Thromb Res 1998; 92: S53-S58. [ Links ]

