










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNefrología (Madrid)
versión On-line ISSN 1989-2284versión impresa ISSN 0211-6995
Nefrología (Madr.) vol.31 no.3 Cantabria 2011
El papel de los inhibidores de mTOR en las enfermedades renales
The role of mTOR inhibitors in renal diseases
J.C. Rodríguez Pérez
Servicio de Nefrología. Hospital Universitario de Gran Canaria Dr. Negrín. Las Palmas de Gran Canaria
Dirección para correspondencia
Introducción
La transducción de señales en los diferentes tipos de células frecuentemente implica la activación, condicional o constitutiva, de receptores tirosina quinasas, los cuales disparan múltiples quinasas citoplasmáticas. Tales rutas de señalización pueden operar independientemente, de forma paralela y/o a través de interconexiones que promueven el desarrollo de diferentes patologías. Entre las rutas de señalización más importantes son la ruta de la fosfatidil inositol 3 quinasa (PI3K), la proteína quinasa C (PKC) y la de la proteína quinasa activada por mitógeno (MAPK/Ras)1.
La proteína mTOR
mTOR (mammalian Target of Rapamycin) es una serina/treonina quinasa de 289 kDa. La familia de proteínas TOR tiene funciones pleiotrópicas y participa en la regulación del inicio de la transcripción del ARNm y la traducción a proteína en respuesta a concentraciones intracelulares de aminoácidos y otros nutrientes esenciales. Interviene en la organización del citoesqueleto de actina, en el tráfico de membrana, en la degradación de proteínas, en la señalización de PKC y en la biogénesis del ribosoma. mTOR regula rutas de señalización esenciales y está implicada en el acoplamiento del estímulo de crecimiento y la progresión del ciclo celular.
Existen dos complejos que contienen mTOR: el complejo sensible a rapamicina (mTORC1), que se define por su interacción con la proteína raptor (regulatory-associated protein of mTOR), y un complejo insensible a rapamicina (mTORC2), que se define por su interacción con rictor (rapamycin-insensitive companion of mTOR). mTOR es una quinasa clave que actúa por debajo de PI3K. Muchas evidencias apoyan la hipótesis de que mTOR es la llave del catabolismo y anabolismo celular que determina si las células, y en particular las células cancerígenas, deben crecer y proliferar. Además, mTOR tiene efectos en la regulación de la apoptosis.
mTOR en la forma de los dos complejos de señalización citados como mTORC1 y mTORC2 con sus dos proteínas diferentes, raptor y rictor, respectivamente, establecen su conexión a dos distintos brazos de acción de la ruta de mTOR. La ruta raptor-mTOR (-mTORC1) regula el crecimiento celular (acumulación de masa celular) y la proliferación a través de p70S6K y 4EBPs. Responde a nutrientes y a factores de crecimiento, en parte debidos a reguladores como TSC1/TSC2 (complejo esclerosis tuberosa 1-hamartina complejo esclerosis tuberosa 2-tuberina) y Rheb (familia Ras de GTPasa). La fosforilación de mTOR (mTORC1) por AKT (también llamada proteína quinasa B-PKB) ocurre a través de la inactivación del complejo de esclerosis tuberosa (TSC) y directamente por Rheb2,3.
El complejo rictor-mTOR (mTORC2) regula AKT/PKB, PKCα, Rho/rac, para controlar la polaridad celular y el citoesqueleto. Los factores de crecimiento y los AA activan AKT y mTOR a través de PI3K1.
Hay factores de transcripción que puedenser activados o ser inhibidos tras la fosforilación de AKT. AKT activa el factor de transcripción NF-kB, lo que tiene como consecuencia un incremento en la transcripción de genes antiapoptóticos. El factor de transcripción NF-kB es el mediador central de la respuesta inmune, de la respuesta inflamatoria y de la respuesta de supervivencia celular. Tras su activación, IKK fosforila IkB, marcándolo para la ubiquitinación y degradación en el proteosoma. Esto expone los lugares de localización nuclear de NF-kB y le permite la traslocación al núcleo donde induce la expresión de genes antiapoptóticos. Los factores de crecimiento, como el factor vascular de crecimiento endotelial (VEGF), activan NF-kB y protegen contra la apoptosis y, por el contrario, la inhibición de NF-kB sensibiliza a la célula a una amplia variedad de estímulos proapoptóticos3.
Papel de mTOR en el fracaso renal agudo
La regeneración y restauración de la morfología y funcionalismo renal dependerá en parte de la capacidad del resto de las células tubulares renales viables para proliferar y restaurar el epitelio dañado4. mTOR es una quinasa ubicua y su inhibición por rapamicina también bloquea la proliferación incluyendo las células en el riñón5. mTOR desempeña un importante papel en el proceso de regeneración y recuperación tras un fracaso renal agudo (FRA) experimental. La actividad mTOR está ausente o es baja en el riñón normal, pero aumenta de forma importante tras un proceso de isquemia-reperfusión. La inhibición de la mTOR por rapamicina retrasa la recuperación-reparación renal5.
Papel de mTOR en la enfermedad renal crónica
La vía mTOR desempeña un importante papel en los mecanismos que intervienen en la progresión de la enfermedad renal crónica (ERC) causada por la diabetes o porotras causas. La rapamicina reduce la inflamación intersticial, la fibrosis y la pérdida de función renal que se asocia con la progresión de la ERC.
Varios estudios han demostrado la importancia de la activación de la mTOR en las formas fisiológicas y patológicas de la hipertrofia renal y otros órganos, incluyendo la hipertrofia de la nefropatía diabética (ND)6. Este fenómeno contribuye al daño podocitario y a la progresiva pérdida de función renal7. Además, se va a producir un fenómeno dependiente de la activación de mTOR consistente en el incremento de síntesis de proteínas de matriz que contribuirá al engrosamiento de la membrana basal glomerular y a la acumulación de matriz mesangial típicos de la ND8. La activación de mTOR en la diabetes es, al menos, en parte provocada por la hiperglucemia vía activación de la AKT8. La rapamicina no sólo se ha visto que reduce la actividad mTOR en modelos in vivo, sino que también disminuye los cambios característicos de la ND ya comentados, asociados con una reducción de la albuminuria9.
En ERC no diabéticas pueden observarse fenómenos similares a los encontrados en la ND, con un aumento en la expresión de citoquinas proinflamatorias y profibróticas, infiltración del intersticio renal por células inflamatorias y fibrosis renal10. El tratamiento con rapamicina en un modelo de glomerulonefritis membranosa en rata reduce todos estos fenómenos11.
Papel de mTOR en la poliquistosis renal autosómica dominante
La poliquistosis renal autosçomica dominante (PQRAD) es una enfermedad genética caracterizada por la formación de quistes múltiples dentro del parénquima renal, lo cual resulta en pérdida de su función. Se calcula que afecta hasta a un caso porcada 400-1.000 recién nacidos. La PQRAD está relacionada con las mutaciones de los genes PKD1 y PKD2, que se localizan en los cromosomas 16 y 4, respectivamente. El gen PKD1 codifica para la proteína de transmembrana policistina 1 (PC1). A esta proteína se le atribuyen funciones como la adhesión célula-célula, la adhesión célula-matriz, la transducción de señales intracelulares y la regulación de la policistina 2 (PC2), en tanto que el gen PKD2 codifica la proteína PC2, que es un canal de calcio2,12. El complejo de PC1 y PC2 es fundamental para el mantenimiento del fenotipo fisiológico de las células epiteliales tubulares.
Los complejos proteicos de policistina son fundamentales para mantener la homeostasis intracelular del calcio. Recientemente, se ha hallado que todas las células epiteliales del túbulo renal, salvo las intercaladas, poseen un único cilio primitivo que tiene funciones mecanorreceptoras y quimorreceptoras. La estimulación de estos cilios aumenta el calcio intracelular a través del complejo de PC1 y PC2. El calcio intracelular, a su vez, controla muchos de los procesos celulares como la proliferación. Por tanto, los fármacos que reducen la formación de adenosín monofosfato cíclico (AMPc) o que aumentan el calcio intracelular podrían representar un tratamiento para la PQRAD. El calcio además, está involucrado en las vías de señalización relacionadas con los factores de crecimiento, activando cascadas de señalización de algunas proteinquinasas. Con ello, la PC1 regula la actividad de mTOR (la pérdida de actividad de PC1 en la PQRAD permite una importante actividad de mTOR en el interior de las células epiteliales de los quistes renales en modelos murinos y humanos), lo que representaría un objetivo terapéutico2,13.
Desarrollo de la rapamicina
La rapamicina, también llamada sirolimus, es un antibiótico natural sintetizado por S. hygroscopicus. Esta bacteria fue descubierta hace 30 años en la tierra de la isla de Pascua, Rapa Nui, de ahí el nombre de rapamicina. Es una lactona desarrollada inicialmente como agente antifúngico. En su forma pura muestra un aspecto de polvo blanco cristalino insoluble en solución acuosa pero soluble en solventes orgánicos. La estructura química se expone en la figura 1.
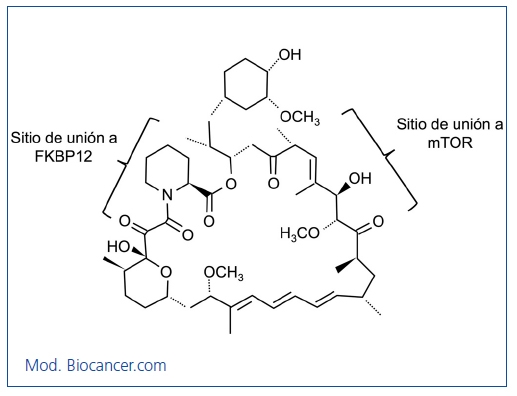
Figura 1. Estructura química de la rapamicina.
Entre 1982 y 1988, la rapamicina se desarrolló como un agente inmunosupresor. Gracias a estos estudios se dilucidó el mecanismo de acción de esta molécula. La rapamicina, a través de su grupo metoxi, interactúa con la proteína de unión a la inmunofilina FK506 (FKBP12). El complejo rapamicina-FKBP12 se fija específicamente a la proteína mTOR inhibiendo las rutas de señalización efectoras dependientes de dicha proteína14. La rapamicina inhibe la proliferación de las células T inducida por antígeno y las respuestas proliferativas inducidas por citoquinas, incluyendo interleuquina 16 (IL-16), immunoglobulin growth factor (IGF), etc. Sigue la vía del citocromo Cyp450 3A4 como el principal sistema responsable de la biotransformación del fármaco, generando metabolitos inactivos.
Se ha demostrado un elevado grado de sinergismo de este fármaco con la ciclosporina15, tanto in vivo como in vitro; de este modo, disminuye la dosis necesaria para una inmunosupresión efectiva, se reduce la probabilidad de rechazo en el trasplante renal y se minimiza la toxicidad inducida por la ciclosporina16.
Un aspecto destacable de la rapamicina como inmunosupresor es la ausencia de efectos secundarios sobre la hemodinámica renal. El tratamiento con rapamicina preserva el filtrado glomerular y el flujo sanguíneo renal tanto en ratas normales como en ratas deplecionadas de sal o con hipertensión espontánea17. El tejido renal parece estar protegido durante el tratamiento con rapamicina a través de la inhibición de la cascada de la angiotensina II intrarrenal. Sin embargo, la rapamicina provoca toxicidad tubular dosis-dependiente en ratas (incluye hipocalcemia e hipofosfatemia), fenómeno que se vincula al retraso en la recuperación de la función epitelial tubular después del daño tisular18.
La rapamicina fue aprobada por la Food and Drug Administration (FDA) de los Estados Unidos en el año 1999 para el tratamiento preventivo del rechazo agudo en combinación con la ciclosporina y los esteroides. Un año más tarde, el fármaco fue aprobado por la agencia europea del medicamento (EMEA) como una alternativa a los antagonistas de la calcineurina en la terapia a largo plazo para evitar el rechazo de trasplantes. Cabe destacar que la rapamicina, al contrario que la ciclosporina, no parece incrementar el riesgo de malignidad sino que más bien disminuye el riesgo de procesos linfoproliferativos posteriores al trasplante (disminuye los niveles deAKT); sin embargo, la rapamicina aumenta los efectos colaterales de la CsA: hipertensión, acné e hirsutismo y también se ha asociado con efectos secundarios levescomo diarrea, taquicardia, edemas, dislipemia y neumonitis no infecciosa.
Además de su capacidad inmunosupresora, se ha demostrado que la rapamicina actúa como agente preventivo sobre la reestenosis de las arterias coronarias19. El hipotético mecanismo responsable de la inhibición de la proliferación de las células del músculo liso vascular por parte de la rapamicina incluye, entre otras, la unión a la inmunofilina FKBP12.
En el artículo que presenta en este mismo número, la Dra. Cabrera, et al.20 comentan los resultados del tratamiento con rapamicina sobre la evolución de los angiomiolipomas en un número nada despreciable considerando su prevalencia, de pacientes afectados de esclerosis tuberosa (ET) o enfermedad de Pringle-Bourneville.
El complejo ET es una enfermedad sistémica con herencia autosómica dominante con una prevalencia de un caso por cada 6.000 nacidos vivos21 caracterizada por tumores benignos (hamartomas) en múltiples órganos y sistemas, incluyendo cerebro, piel, riñones, pulmones, corazón y retina. Los angiomiolipomas son tumores ricos en tejido adiposo, músculo y vasos sanguíneos que pueden sangrar o infiltrar los riñones provocando deterioro de función renal hasta en el 80% de los pacientes22.
Las mutaciones que pueden ocurrir en cualquiera de los dos genes de ET, TSC1 (hamartina) o TSC2 (tuberina) se encuentran por encima del 85% de los pacientes con ET23. Las proteínas codificadas por estos dos genes forman un complejo supresor de tumores actuando a través de una vía homóloga de Ras enriquecida en una proteína cerebral (Rheb) limitando la activación de mTOR (mTORC1). Cuando TSC1 o TSC2 son deficitarios, mTORC1 se sobreexpresa de forma constitutiva, lo que provocará un crecimiento celular, proliferación y síntesis proteica anormalmente alta24.
En el ensayo clínico en fase IV que presentan los citados autores20, se demuestra una reducción significativa del volumen de los angiomiolipomas ya desde los seis meses de tratamiento con rapamicina (un 55,1% de media), alcanzando una reducción del 66,3% a los 12 meses. Esta reducción del volumen de los angiomiolipomas parece deberse al efecto de inhibición de la mTOR y a su efecto sobre el VEGF. Otro aspecto de gran interés que expresan los autores en el citado trabajo es la posibilidad de reducir las dosis de rapamicina tras conseguir la máxima reducción del volumen de los angiomiolipomas, que parece situarse entre los 12 y los 24 meses20,25, ya que la retirada completa del tratamiento favorecería la recuperación del volumen de este tipo de tumores25,26. Queda por conocer si el efecto de rapamicina sería uniforme en todo tipo de angiomiolipoma sea cual fuera su dimensión y localización (unilateral o bilateral). De igual forma, al no existir estudio genético, ya que el fenotipo puede ser diferente si la mutación se localiza en TSC1 o TSC2, no sabemos si la respuesta a rapamicina pudiera variar.
Sólo un paciente fue excluido antes de los 12 meses por reactivación de un eritema nudoso. No se objetivaron cambios en la función renal considerando que se mantuvieron niveles plasmáticos constantes de rapamicina. A pesar de los efectos adversos referidos en la bibliografía médica25, los autores encuentran una mayor incidencia de aftas orales y dislipemia20. Es de destacar la disminución de tamaño y el menor realce de los angiofibromas faciales en estos pacientes.
Existen muy pocos datos en la bibliografía en este sentido, datos aislados sobre algún caso clínico26 y un ensayo clínico publicado en New England Journal of Medicine con 25 pacientes de los que sólo 20 cumplieron los 12 meses de seguimiento25. Ninguno de ellos aporta estudio genético. Aunque como citan los autores en su artículo20 existen varios estudios en marcha en Estados Unidos y Europa.
La rapamicina, a través de la inhibición de la mTOR, es una alternativa terapéutica en esta enfermedad. Se necesitan más estudios para definir los riesgos y beneficios de rapamicina a largo plazo en este tipo de enfermedades hereditarias, su uso en monoterapia o en asociación y según localización y tamaño de los angiomiolipomas.
Referencias Bibliográficas
1. Pérez Machín R, Rodríguez Díaz Y, Vega Hernández MC. La ruta mTOR como diana terapéutica. BioCancer 2006;3. [ Links ]
2. Masoumi A, Reed Gitomer B, Kelleher C, Schrier RW. Potential pharmacological interventions in polycystic kidney disease. Drugs 2007;67:2495-510. [ Links ]
3. Lieberthal W, Levine JS. The role of the mammalian target of rapamycin (mTOR) in renal disease. J Am Soc Nephrol 2009;20:2493-502. [ Links ]
4. Bonventre JV. Dedifferentiation and proliferation of surviving epithelial cells in acute renal failure. J Am Soc Nephrol 2003;14(Suppl 1):S55-S61. [ Links ]
5. Lieberthal W, Fuhro R, Andry CC, Rennke H, Abernathy VE, Koh JS, et al. Rapamycin impairs recovery from acute renal failure: role of cell-cycle arrest and apoptosis of tubular cells. Am J Physiol Renal Physiol 2001;281:F693-F706. [ Links ]
6. Lee CH, Inoki K, Guan KL. mTOR pathway as a target in tissue hypertrophy. Annu Rev Pharmacol Toxicol 2007;47:443-67. [ Links ]
7. Hostetter TH. Hyperfiltration and glomerulosclerosis. Semin Nephrol 2003;23:194-9. [ Links ]
8. Kasinath BS, Mariappan MM, Sataranatarajan K, Lee MJ, Feliers D. mRNA translation: Unexplored territory in renal science. J Am Soc Nephrol 2006;17:3281-92. [ Links ]
9. Lloberas N, Cruzado JM, Franquesa M, Herrero-Fresneda I, Torras J, Alperovich G, et al. Mammalian target of rapamycin pathway blockade slows progression of diabetic kidney disease in rats. J Am Soc Nephrol 2006;17:1395-404. [ Links ]
10. Eddy AA, Neilson EG. Chronic kidney disease progression. J Am Soc Nephrol 2006;17:2964-6. [ Links ]
11. Bonegio RG, Fuhro R, Wang Z, Valeri CR, Andry C, Salant DJ, et al. Rapamycin ameliorates proteinuria-associated tubulointerstitial inflammation and fibrosis in experimental membranous nephropathy. J Am Soc Nephrol 2005;16:2063-72. [ Links ]
12. Aguiari G, Trimi V, Bogo M, Mangolini A, Szabadkai G, Pinton P, et al. Novel role for polycystin-1 in modulating cell proliferation through calcium oscillations in kidney cells. Cell Prolif 2008;41:554-73. [ Links ]
13. Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 2006;103:5466-71. [ Links ]
14. Baker H, Sidorowicz A, Sehgal SN, Vezina C. Rapamycin (AY-22,989), a new antifungal antibiotic. III. In vitro and in vivo evaluation. J Antibiot (Tokyo) 1978;31:539-45. [ Links ]
15. Davies CB, Madden RL, Alexander JW. Effect of a short course of rapamycin, cyclosporin A, and donor-specific transfusion on rat cardiac allograft survival. Transplantation 1993;55:1107-12. [ Links ]
16. Trepanier DJ, Gallant H, Legatt DF, Yatscoff RW. Rapamycin: distribution, pharmacokinetics and therapeutic range investigations: an update. Clin Biochem 1998;31:345-51. [ Links ]
17. DiJoseph JF, Mihatsch MJ, Sehgal SN. Renal effects of rapamycin in the spontaneously hypertensive rat. Transpl Int 1994;7:83-8. [ Links ]
18. Andoh TF, Burdmann EA, Fransechini N, Houghton DC, Bennett WM. Comparison of acute rapamycin nephrotoxicity with cyclosporine and FK506. Kidney Int 1996;50:1110-7. [ Links ]
19. Sousa JE, Sousa AG, Costa MA, Abizaid AC, Feres F. Use of rapamycin-impregnated stents in coronary arteries. Transplant Proc 2003;35:165S-170S. [ Links ]
20. Cabrera López C, Martí T, Catalá V, Torres F, Mateu S, Ballarín Castán J, et al. Efectos de la rapamicina en los angiomiolipomas de pacientes con esclerosis tuberosa. Nefrologia 2011;31(3):292-8. [ Links ]
21. Krueger DA, Franz DN. Current Management of tuberous sclerosis complex. Paediatr Drugs 2008;10:299-313. [ Links ]
22. Bissler JJ, Kingswood JC. Renal angiomyolipomata. Kidney Int 2004;66:924-34. [ Links ]
23. Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med 2006;355:1345-56. [ Links ]
24. Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J 2008;412:179-90. [ Links ]
25. Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 2008; 358:140-51. [ Links ]
26. Wienecke R, Facler I, Linsenmaier U, Mayer K, Licht T, Kretzler M. Antitumoral activity of rapamycin in renal angiomyolipoma associated with tuberous sclerosis complex. Am J Kidney Dis 2006;48:E27-E29. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
José Carlos Rodríguez Pérez,
Servicio de Nefrología,
Hospital Universitario de Gran Canaria Dr. Negrín,
Bco. La Ballena, s/n,
35010, Las Palmas de Gran Canaria
E-mail:
jrodperd@gobiernodecanarias.org
Enviado a Revisar: 26 Abr. 2011
Aceptado el: 26 Abr. 2011

