










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCCIÓN
En los países industrializados, algunas deficiencias vitamínicas están casi erradicadas (escorbuto, beriberi, pelagra). Si se presentan, se debe a dietas demasiado estrictas o patología médica subyacente. Sin embargo, hay otras que mantienen una incidencia significativa, como el síndrome de Wernicke y la anemia perniciosa, y colocan a los déficits vitamínicos entre las enfermedades neurológicas carenciales más frecuentes.
En esta revisión, abordaremos las enfermedades neurológicas más relevantes en relación con el déficit de vitaminas del grupo B, centrándonos, dentro de estas, en la tiamina, el folato y la cobalamina. La interrelación entre ellas es estrecha: realizan un "trabajo en equipo" que puede dar lugar a situaciones clínicas peculiares, como, por ejemplo, el hecho de que el déficit de folato pueda mostrar un falso déficit de cobalamina, o la absorción de B1 se vea favorecida por la presencia de folato. Intercalaremos la revisión de estas vitaminas con un caso clínico ilustrativo.
CASO CLÍNICO
- Varón de 70 años con antecedentes de úlcera péptica con duodenitis, estenosis duodenal y un fitobezoar (acúmulo de sustancias vegetales en el estómago) en seguimiento.
- A los 67 años ingresó por una pericarditis, con derrame pericárdico severo y fibrilación auricular. Durante el ingreso presentó confusión y marcha inestable, que mejoraron espontáneamente. Quedó mínima alteración de la marcha.
- Acude a Urgencias por empeoramiento de la marcha las últimas semanas. La exploración, compatible con trastorno de cordones medulares posteriores (alteración de sensibilidad propioceptiva) y el hallazgo analítico de folato en sangre de 1,8 ng/mL (déficit si < 2,5), lleva a diagnóstico de degeneración combinada subaguda por déficit de folato. Se cursa el alta en tratamiento con fólico.
DÉFICIT DE COBALAMINA (B12)
FUENTE E INGESTAS RECOMENDADAS(1,2)
Podemos encontrarla en carnes, lácteos, huevos y, con concentraciones mayores, en almejas e hígado 3.
Se recomiendan ingestas de 0,5 a 3 μg al día (mínimo en lactantes, máximo en mayores de 70 años). La reserva total de B12 en el organismo es amplia (de 2-5 mg), aproximadamente la mitad en el hígado, lo que explica que el déficit pueda tardar en ser sintomático 1-3 años.
ABSORCIÓN
En los alimentos está unida a proteínas, también en la saliva (R-binders). Se escinde de las proteínas en el entorno ácido del estómago gracias a la pepsina. Las células parietales gástricas producen factor intrínseco (FI), que se unirá a la B12 en el duodeno mediante las proteasas pancreáticas y ante pH mayor. El complejo FI- B12 es recibido por receptores aún desconocidos en el íleon 4,5. La B12 se exporta a la sangre gracias a una proteína ABC (ATP-binding cassette) y ABCC1 (también llamada proteína de multirresistencia a fármacos) 6. Se une en la sangre a transcobalaminas, TC-I y TC-II (la fisiológicamente más importante), y con ellas llega a las células mediante endocitosis mediada por receptor.
METABOLISMO Y MECANISMO DE ACCIÓN
sus dos formas activas: metilcobalamina y adenosilcobalamina. Actúa como cofactor en los procesos enzimáticos que llevan a la síntesis de ADN en las células en división. Es cofactor para las enzimas metil-malonil-coenzima A mutasa y metionina sintetasa. Así, participa en el paso de ácido metilmalónico-coA a succinil-coenzima-A y de homocisteína a metionina.
Es necesaria para la formación de eritrocitos, la función neurológica (mielinización inicial, desarrollo y mantenimiento de la mielina) y síntesis de ADN.
CAUSAS DE DÉFICIT
El déficit moderado suele ser por déficit de ingesta de alimentos con B12 biodisponible o por desnutrición. Si el problema es absortivo, puede corresponder a las fases gástrica o ileal.
La anemia perniciosa autoinmune es la causa más frecuente de malabsorción. Habrá un déficit de factor intrínseco por lesión de las células parietales gástricas en el contexto de una gastritis autoinmune o tras una gastrectomía.
Otras causas de malabsorción 7 son: resección de íleo terminal, síndrome de asa ciega, cirugía bariátrica, tratamientos con omeprazol o anti-H2 (aclorhidria), enfermedades celíacas, inflamatoria intestinal y pancreáticas.
También puede deberse a la exposición a fármacos antiepilépticos y óxido nitroso. Este último se usa como anestésico y como droga de abuso, y altera el core de cobalto de la cobalamina (el cobalto da el nombre a la vitamina). Una sola exposición puede provocar síntomas neurológicos rápidamente, pero los síntomas pueden retrasarse hasta dos meses. Hay varios casos descritos en dentistas 8.
PATOLOGÍA ASOCIADA
El 2-15% de los ancianos tiene déficit de B12 7. La incidencia de la gastritis atrófica aumenta con la edad. El déficit moderado suele provocar trastorno neurocognitivo y el severo, anemia perniciosa.
Degeneración combinada subaguda
Desmielinización y destrucción axonal, tanto medular como encefálica. Cuando el daño es amplio e incluye desmielinización de nervios craneales, periféricos y del cerebro, se denomina degeneración combinada de sistemas 7. El cuadro clínico tiene un inicio insidioso con parestesias en manos y pies; posteriormente se añadirán debilidad e inestabilidad en la marcha. Estos síntomas se asociarán y a la exploración obtendremos datos de polineuropatía (por lo general leve y de predominio axonal) y mielopatía (lesión de cordones posteriores).
Pueden añadirse otros síntomas: lentitud mental, depresión, confusión, ideación delirante, alucinaciones. En el estudio de un paciente con demencia deberá descartarse déficit de B12.
Pruebas complementarias
- Resonancia magnética (RM) craneal/medular: mielopatía: lesiones en regiones laterales y posteriores medulares. Encefalopatía: múltiples lesiones de sustancia blanca profunda.
- Electromiograma (EMG): polineuropatía axonal.
- Potenciales evocados: somatosensoriales: vías propioceptivas enlentecidas. Visuales: patológicos con frecuencia.
- La prueba principal de despistaje es la cuantificación de B12 en la sangre (niveles normales: 200-900 pg/mL). No son raros los falsos negativos (déficit real, analítica normal) ante hepatopatías y síndromes mieloproloferativos o los falsos positivos (niveles falsamente bajos) ante embarazo, toma de anticonceptivos o déficit de folato. En clínica deben solicitarse ambas vitaminas simultáneamente. Ante una fuerte sospecha de déficit que no se confirma con la analítica, la elevación en suero de metilmalónico y homocisteína indicaría déficit intracelular de B12.
- Si el déficit de B12 compromete a las series hematológicas y sospechamos una anemia perniciosa autoinmune, deberemos buscar: en el hemograma macrocitosis e hipersegmentación de polimorfonucleares sin/con anemia, y en suero, en el 60-90% de los pacientes presencia de anticuerpos anti célula parietal gástrica y anti factor intrínseco (los primeros más sensibles y menos específicos), y de anticuerpos antigastrina, que tienen sensibilidad y especificidad del 70% para anemia perniciosa.
Tratamiento
Cianocobalamina (también hidroxicobalamina): dosis intramuscular de 100 μg al día o 1.000 μg dos veces por semana durante dos semanas. Posteriormente, 1.000 μg semanales durante 2-3 meses. Los niveles de homocisteína y metilmalónico son un buen monitor de respuesta al tratamiento, y se normalizan en 10-14 días.
Pronóstico
Cuanto antes se diagnostique el déficit y se inicie el tratamiento, mejor será el pronóstico. La anemia se resuelve en 1-2 meses, los síntomas neurológicos se estabilizan y pueden mejorar, por lo general de forma parcial, en 6-12 meses. La mejoría radiológica, aunque puede retrasarse meses, con frecuencia se adelanta a la clínica, e incluso puede ser más marcada que esta.
DÉFICIT DE FOLATO (B9)
El déficit de folato es uno de los trastornos nutricionales mundialmente más comunes 7.
FUENTE E INGESTA RECOMENDADAS
Se encuentra en muchas plantas, especialmente de hoja verde, y en el hígado 9. La forma sintética es el ácido fólico, un monoglutamato oxidado más estable, pero la mitad de biodisponible que el natural.
Se recomiendan ingestas entre 70 μg al día en niños y 600 μg en la segunda mitad del embarazo 1. Se ha popularizado reforzar con fólico cereales, harinas y granos para reducir el riesgo de defectos del tubo neural 10.
ABSORCIÓN
Se absorbe en el duodeno y el yeyuno de forma pasiva, y en mayor medida mediante un transportador. Las reservas de folato son moderadas (0,5 a 20 mg); si no se recibe, el déficit analítico comenzará en semanas y el clínico, en meses.
METABOLISMO Y MECANISMO DE ACCIÓN
Están íntimamente ligados al de la cobalamina. Para ser efectivo, el folato tiene que ser reducido a dihidrofolato y, después, a tetrahidrofolato (THF). Este pasa primero a 5,10-metileno THF, y después a L-5-metil THF, que predomina en suero y es captado rápidamente por los hepatocitos y otras células mediante la metilenotetrahidrofolato reductasa. Entra en la célula mediante el receptor multiligando megalina (relacionado con el colesterol LDL y también receptor de otras proteínas).
Una vez en la célula vuelve a ser poliglutamato como forma activa incapaz de salir de la célula 11.
La cobalamina es cofactor en el paso de 5-metil-THF a THF, involucrado en la síntesis de purina, timidina y aminoácidos. La síntesis de THF está emparejada al paso de homocisteina a metionina 9.
CAUSAS DE DÉFICIT
El origen farmacológico es frecuente. Puede producirse déficit ante la toma de antiepilépticos "clásicos" (fenitoína, fenobarbital, carbamazepina) u otros fármacos: inhibidores de la bomba de protones, Ranitidina, Aminopterina, Pirimetamina, Trimetroprim, Triamterene, anovulatorios y, sobre todo, inmunosupresores como la Salazopirina, la Azatioprina y el Metotrexato 12. Otras causas adquiridas: malnutrición, alcoholismo, aumento no satisfecho de los requerimientos (embarazo, lactancia, anemia hemolítica crónica), cirugías gástricas y enfermedades intestinales. También el origen puede ser genético: deficiencia en homocigosis de cistationina-β-sintetasa, mutación C-677-T en el gen de la metilentetrahidrofolato-reductasa 8.
DIAGNÓSTICO(9)
Se considera el déficit franco con niveles séricos inferiores a 2,5 μg/l, si bien el folato eritrocitario es más informativo de la situación intracelular. La homocisteína está elevada en el 86% de los pacientes con déficit significativo de folato.
PATOLOGÍA ASOCIADA
Similar al déficit de B12, pero es menos frecuente la clínica neurológica. La presencia de folato bajo y homocisteína elevada se considera un factor de riesgo de demencia (incluyendo enfermedad de Alzheimer) y depresión.
Se calcula que el 50 % de los defectos del tubo neural se deben a déficit de folato en la madre alrededor de la concepción 9. Los suplementos de fólico durante el embarazo pueden reducir el riesgo, aunque no haya déficit previo, pero no lo eliminan del todo.
TRATAMIENTO(9)
Ácido fólico: 1 mg vo C/8 h 4 semanas, seguido de 1 mg diario (factible por vía parenteral si daño agudo o malabsorción). En el embarazo se recomiendan suplementos de 0,4 mg al día, o dosis mayores en presencia de antiepilépticos.
Ante déficit de folato y B12, si se administra solamente folato puede empeorar, primero, el cuadro neurológico y luego, el psiquiátrico. Insistimos en que siempre debe conocerse también el nivel de B12.
Y NUESTRO PACIENTE...
Al mes vuelve a Urgencias. Se añaden al cuadro clínico insuficiencia cardiaca derecha y derrame pericárdico moderado. Diagnóstico: pericarditis. Tratamiento: Furosemida.
Evolución: mejoría de los síntomas cardiológicos inicialmente, pero no de la marcha inestable. Unos días después empeora, con nuevos síntomas: síndrome vertiginoso, nistagmo horizontal en ambas posiciones laterales de la mirada sin oftalmoparesias, ataxia de tronco, dismetría, tetraparesia e hipoestesia en "guante y calcetín".
DÉFICIT DE TIAMINA (B1)
La tiamina es el precursor de la forma más activa, la tiamina pirofosfato.
FUENTE E INGESTAS RECOMENDADAS
Se adquiere de cereales sin refinar, germen de trigo, levadura, harina de soja y carne de cerdo. Ingesta recomendada 1: entre 0,3 mg al día en niños y 1,5 mg en la segunda mitad del embarazo.
ABSORCIÓN
Por transportador activo si la ingesta es mínima y por difusión pasiva si "sobra" en el yeyuno y el íleon 13. Se desfosforila para atravesar la mucosa hacia el torrente sanguíneo mediante la bomba de sodio dependiente de ATP, y vuelve a fosforilarse dentro de la célula, salvo en los eritrocitos, en los que entra por difusión pasiva 14.
Es preciso garantizar el aporte continuo 15, ya que la vida media es de 10-20 días. El almacenamiento tisular (sobre todo en músculo esquelético, hígado, corazón, riñones y cerebro) es escaso, y la absorción de tiamina es limitada y aumenta poco si se aporta al individuo grandes cantidades de B1 (no así el folato).
METABOLISMO Y MECANISMO DE ACCIÓN
Actúa como coenzima en el metabolismo de carbohidratos, lípidos y aminoácidos de cadena ramificada. También en la síntesis de mielina. Además, tiene un papel no identificado (aunque no como coenzima) en el inicio de la propagación del impulso nervioso 14, posiblemente en la transmisión colinérgica y serotoninérgica a través de efecto sobre el canal de sodio 9.
La demanda de tiamina pirofosfato depende del consumo de glucosa: esta, por vía venosa, puede dar lugar a patología severa en pacientes con déficit latente de tiamina (necesaria para la oxidación de la glucosa).
CAUSAS DEL DÉFICIT(16)
Aunque no exclusiva, la causa más frecuente es el alcoholismo, pues en estos pacientes se unen: aporte inadecuado, absorción gastrointestinal reducida, conversión disminuida a metabolitos activos, aumento de demanda por el metabolismo de carbohidratos y reducción del almacenamiento hepático. Otras causas: malnutrición de otro origen, vómitos repetidos, enfermedades sistémicas graves, cirugía bariátrica, fármacos antiácidos, alimentación parenteral prolongada y requerimientos aumentados: embarazo, enfermedad crítica, malignidad (y tratamientos quimioterápicos), hipertiroidismo, infección y administración de carbohidratos por vena.
PATOLOGÍA ASOCIADA
Se producirá por acúmulo de ácido láctico y déficit de la captación de oxígeno celular. Al tratarse de alteraciones en el metabolismo de la glucosa, el sistema nervioso es el más expuesto (alteración de los gradientes osmóticos, acúmulo de glutamato, alteración de la permeabilidad de la barrera hematoencefálica).
Síndrome de Wernicke-Korsakoff (SWK)
Descrito en 1881 por Karl Wernicke. Poco después, Korsakoff describió un trastorno de la memoria a corto plazo en alcohólicos. La vinculación de los síntomas y los hallazgos neuropatológicos de ambos autores permitía asumir que estos pacientes tenían algo en común, pero hasta mediados del siglo xx no se supo que era el déficit de tiamina 16.
No se trata de una entidad rara: en autopsias se ven lesiones sugestivas en el 0,8-2,8% de los pacientes, y hasta el 12% en casos de alcoholismo. Se diagnostica con mucha menor frecuencia, pero probablemente porque se nos escapan cuadros incompletos o porque achacamos los síntomas a otras enfermedades del paciente (uremia, sepsis, etc.) 17.
Las características clínicas son muy variadas. Es frecuente la afectación bilateral del nervio motor ocular externo, pero pueden añadirse otras oftalmoparesias, nistagmo horizontal o vertical, desviaciones de la mirada, anisocoria y ataxia axial. Con menor frecuencia encontraremos polineuropatía.
Aproximadamente el 80% de los pacientes desarrollan de forma residual un síndrome de Korsakoff: cuadro amnésico característico con afectación de la memoria de trabajo y amnesia anterógrada. Desarrollan una demencia en la que son características las confabulaciones y frecuentes, la confusión y la desorientación 10.
El diagnóstico del SWK es clínico, pues no disponemos de pruebas complementarias que lo confirmen sin que retrasemos el tratamiento. La tiamina sérica o urinaria no reflejan la concentración de tiamina tisular. Son más fiables los niveles de tiamina eritrocitaria y la actividad de la transcetolasa. También puede haber aumento de ácido láctico.
En algo más de la mitad de los pacientes la resonancia magnética en T2 y FLAIR muestra hiperseñales talámicas, periacueductales y en el techo del IV ventrículo.
El tratamiento consiste en el aporte de tiamina 300 mg endovenosa al día la primera semana, posteriormente 100 mg diarios y, si hay riesgo de déficit, 50-100 mg al día vía oral de forma indefinida. Debe corregirse el déficit de magnesio, que interviene en la conversión de tiamina en su forma activa de tiamina pirofosfato, y su déficit es causa de mala respuesta a la tiamina 18.
En toda situación de riesgo potencial o de sospecha de SWK hay que administrar, sin demora, tiamina por vía parenteral, y siempre antes que la glucosa.
¿Qué obtendremos del tratamiento? La oftalmoparesia mejora y pronto, también el síndrome confusional agudo; la ataxia y la neuropatía, si mejoran, tardan más; la amnesia no suele resolverse.
FINAL DEL CASO CLÍNICO Y CONCLUSIÓN
Nuevos síntomas, nuevo estudio:
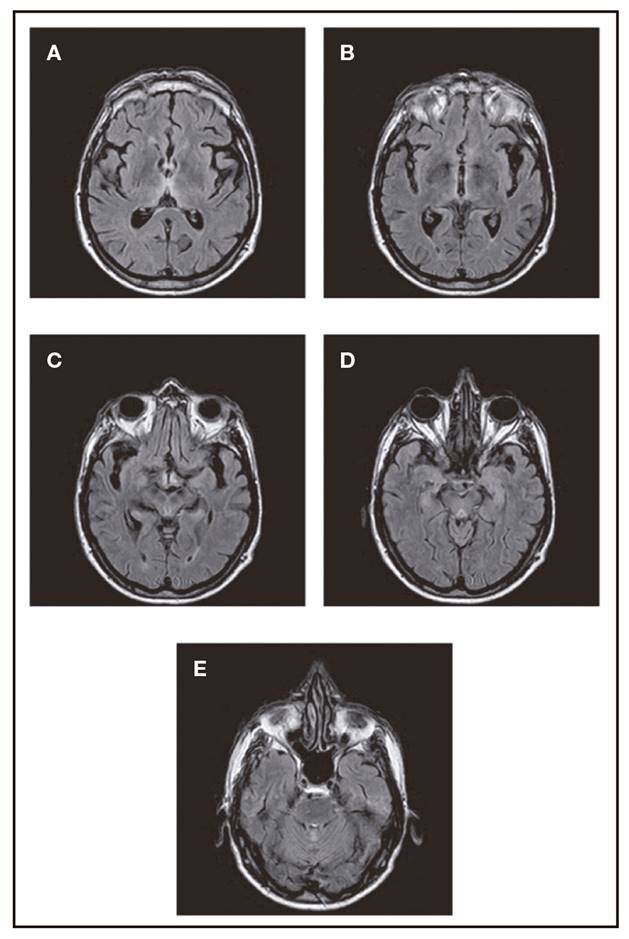
Figura 1. RM craneal del paciente. A y B. Hiperintensidades en T2, FLAIR y difusión en la región medial de ambos cuerpos talámicos. C y D. Sustancia gris subependimaria tanto alrededor del tercer ventrículo como periacueductal. E. Vermix cerebeloso.
Ante la gravedad del cuadro, y tras descartar otras causas de pericarditis, optamos por considerar que se trataba de un proceso sistémico disinmune o carencial, y se inició tratamiento simultáneamente con inmunoglobulina humana inespecífica y tiamina. A las 48 h de tratamiento, la mejoría era marcada. La RM craneal, que mostraba las alteraciones características del SWK, a las dos semanas se había normalizado. Alta al mes del ingreso, con mínima tetraparesia, hipoestesia en guante y calcetín y ataxia moderada (precisaba de andador). A los cinco meses había podido cambiar el andador por un bastón. No quedaron secuelas cognitivas.
¿Qué había ocurrido? Al no tener antecedentes de alcoholismo, el SWK no era la opción más probable, en un principio. El fitobezoar llevó al déficit de absorción de tiamina y folato, y esto a polineuropatía, más dependiente del folato, y por el déficit de tiamina, a beriberi húmedo y SWK. Empeoró en el segundo ingreso porque los diuréticos (para el tratamiento de la insuficiencia cardiaca) aumentan el déficit de tiamina.
Con este caso queremos mostrar la importancia de "tener en mente" las manifestaciones neurológicas por déficit vitamínicos para poder llegar al diagnóstico cuanto antes y, de este modo, convertir el tratamiento en curación.

