










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.19 no.4 abr. 2002
REVISIÓN DE CONJUNTO
Genética de las sobrecargas férricas
A. Zúñiga Cabrera, M. A. Orera Clemente*
Laboratorio de Biología Molecular. Hospital de la Ribera. Alzira. Valencia.
*Unidad de Genética. Hospital General Gregorio Marañón. Madrid
Genetic aspects of iron overloads
Trabajo aceptado: 5 de julio de 2001
Correspondencia: Ángel Zúñiga Cabrera. Lab. Biología Molecular. Hospital de la Ribera. Alzira. Valencia.
INTRODUCCIÓN
La hemocromatosis hereditaria (HC) es la enfermedad genética más frecuente con una incidencia de 1 por cada 200-400 individuos en las poblaciones de origen caucásico. El carácter génico de la enfermedad fue controvertido durante muchos años y no quedó aclarado hasta que se determinó su asociación con los antígenos HLA A3 y B14 (1). Durante casi 20 años se buscó el gen en esta región hasta que en 1996 se identificó el gen HFE o HLA-H mediante clonaje posicional (2).
La proteína HFE tiene un papel fundamental en la homeostasis del hierro, modulando la interacción entre la transferrina y su receptor, con la formación de un complejo en el que interviene la β2-microglobulina. Las mutaciones del gen HFE impiden la ubicación correcta de la proteína HFE en la membrana o impiden su asociación con β2-microglobulina. Aunque el mecanismo exacto de acción se desconoce, el resultado final es un aumento en la absorción intestinal y la acumulación de Fe intracelular.
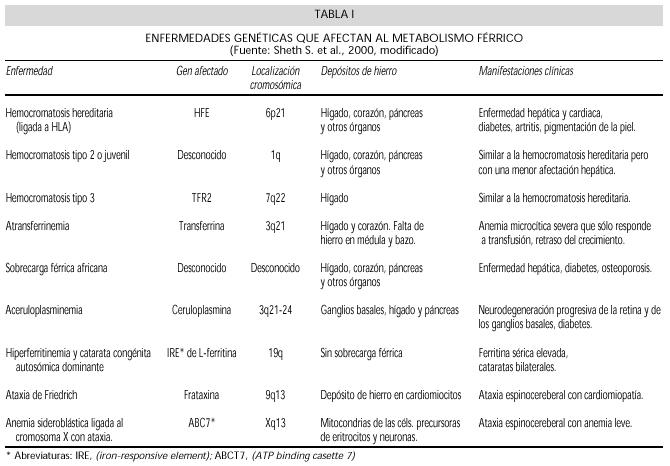
Las alteraciones de la homeostasis del Fe son numerosas y frecuentes, originando tanto situaciones de deficiencia como de sobrecarga. Además de la HC existen situaciones de sobrecarga férrica, causadas por las alteraciones de otras proteínas. Estas enfermedades se enumeran en la tabla I.
PROTEÍNAS QUE INTERVIENEN EN EL METABOLISMO DEL HIERRO
En los últimos años se ha producido un avance importante en el descubrimiento de los mecanismos implicados en el metabolismo férrico, describiéndose las relaciones existentes con el sistema inmune y el sistema nervioso, y señalándose la relación con el metabolismo del cobre. A continuación se detallan las proteínas involucradas en la homeostasis férrica, desde su entrada en el organismo hasta su utilización y almacenamiento celular (Fig. 1).

Absorción intestinal de hierro
El transportador de metales divalentes-1 (DMT1) es una proteína de membrana de una sola cadena que tiene 12 regiones transmembranosas. Interviene en el transporte de hierro desde la luz intestinal al enterocito y en el resto de las células interviene en al paso del endosoma al citoplasma (3). La proteína HFE se localiza en la región perinuclear de las células epiteliales de las criptas intestinales, por lo que se predice una función a este nivel todavía sin determinar.
Entrada en la circulación sistémica
En este paso intervienen "exportadores" de hierro como la hefestina y la ferroportina-1. La hefestina es una proteína análoga a la ceruloplasmina que se encuentra en la membrana del enterocito. Se ha localizado en el brazo largo del cromosoma X (Xq11-12). Los ratones sla con deficiencia en hefestina padecen anemia hipocrómica y son capaces de absorber Fe de la luz intestinal, pero tienen disminuida la excreción al intestino. La consecuencia de ambos efectos es la acumulación de Fe en el enterocito (4).
Transporte plasmático
Realizado por la transferrina (TF) que es una glicoproteína sintetizada en el hígado con dos lugares de unión para el hierro. Interviene también en el transporte de Fe en el fluido extracelular. El gen se encuentra localizado en 3q21 (5). La ausencia de transferrina detectable en plasma constituye la atransferrinemia.
Entrada en la célula
Los mecanismos de entrada de Fe en las células presentan algunas diferencias dependiendo de la actividad biológica de cada una. Se ha visto como en el enterocito la entrada está mediada por el DMT1. El resto de células utilizan un sistema de transporte específico constituido por los receptores de transferrina: TfR y TfR2. En general los TfR intervienen en la entrada de holotransferrina a la célula y en la liberación del hierro de la TF que sucede intracelularmente. Los TfR se encuentran en la superficie de todas las células nucleadas, en mayor o menor número dependiendo del requerimiento celular de Fe. El receptor tradicional se encuentra ubicado en 3q29 (6). Se ha descrito un análogo del receptor (receptor de transferrina 2) que se localiza en el brazo largo del cromosoma 7 (7q22). El TfR 2, que también se une a la TF, tiene la misma función que TfR y se expresa predominantemente en el hígado (7).
La unión entre la TF y su receptor está modulada por la interacción de la proteína HFE y la β2-microglobulina (8). En condiciones normales el heterodímero HFE-β2-microglobulina forma un complejo estable con el receptor de transferrina , disminuyendo la afinidad del receptor por la transferrina (9). Cuando la proteína HFE no es funcional, se elimina este control negativo de modo que tiene lugar una incorporación de hierro a la célula de manera no regulada. En sistemas modelo en los que se ha eliminado el gen HFE (ratones knock-out), se observa una acusada sobrecarga férrica (10). Este hecho se considera la prueba definitiva de que el gen HFE es responsable de la HC y de que el fenotipo de la HC es debido a la pérdida de la función de la proteína HFE.
El gen HFE codifica una proteína transmembranosa de 343 aminoácidos que posee dos dominios extracelulares funcionales: una región con dos hélices alfa (α1 y α2) y un dominio tipo inmunoglobulina que se une a la β2-microglobulina (α3). Se encuentra localizado en el brazo corto del cromosoma 6 (6p21) y es una glicoproteína del tipo MHC. Este gen está mutado en la mayoría de los pacientes afectos de HC hereditaria.
El complejo hierro-transferrina-receptor de transferrina-proteína HFE se introduce en la célula por vía endosómica. La acidificación del endosoma produce la liberación de hierro del complejo, y el paso al citoplasma con la intervención de DMT1. El hierro así liberado puede incorporarse a compuestos metabólicos activos o bien almacenarse.
La proteína HFE parece estar implicada también en la absorción intestinal y en la entrada a la circulación del Fe, aunque los mecanismos de acción de la proteína a estos niveles no están claramente definidos (11).
Existe otra proteína denominada Estimulador del Transporte de Hierro (SFT), que permite el paso a la célula del Hierro libre y del Fe unido a transferrina en los cultivos celulares de hepatocitos. Los niveles de SFT parecen estar directamente relacionados con los niveles intracelulares de hierro, aunque por el momento se desconoce su relación con el resto de proteínas implicadas (12).
Almacenamiento intracelular
El hierro se almacena en el citoplasma unido a la ferritina, de manera que se consigue una doble función: detoxificación y disponibilidad inmediata de hierro intracelular. Cada molécula de ferritina consta de 24 subunidades de dos tipos H (pesadas) y L (ligeras). La subunidad H tiene un peso molecular de 21.100, está codificada en un gen localizado en 11q12, y tiene un sitio de unión para la ceruloplasmina. La subunidad L tiene un peso de 19.700 está codificada por un gen que se localiza en 19q13 y no tiene sitio de unión de la ceruloplasmina. La subunidad H está implicada en la oxidación del hierro ferroso y la subunidad L está implicada en la formación del núcleo de hierro de la ferritina (13) La ausencia de subunidad H produce la hiperferritinemia con catarata congénita (14).
La ceruloplasmina es una glicoproteína que contiene el 95% del cobre encontrado en el plasma. Se piensa que es la ferroxidasa que produce el paso de Fe2+ a Fe3+, y que no tiene ningún papel directo en el transporte del cobre. Está codificada por un gen que se encuentra en 3q21-24 cuyas mutaciones dan lugar a la aceruloplasminemia (15).
La disponibilidad de hierro intracelular está controlada por las proteínas reguladoras de hierro: IRP-1 e IRP-2 que son represores traducionales de la síntesis del receptor de transferrina, DMT1 y de ferritina. En el citoplasma, las proteínas IRP se unen a una región específica de los RNA mensajeros que reciben el nombre de IRE (elementos con respuesta al hierro) (16). Ambas proteínas disminuyen la tasa de síntesis de los genes encargados de la absorción, transporte y almacenamiento de Fe. El aumento de Fe intracelular induce un aumento de IRP-1 de baja afinidad por el RNA, disminuyendo la capacidad de unión entre IRP-1 y los IRE mientras que la disminución del hierro intracelular propicia la producción de IRP-1 de alta afinidad por el RNA. De esta forma se establece una conexión entre la cantidad de hierro intracelular disponible y las necesidades de hierro celulares (17).
Metabolismo mitocondrial del Fe
Se ha sugerido la existencia de un ciclo mitocondrial del hierro que implicaría la existencia de un proceso regulado de entrada, control del almacenamiento de Fe, y un mecanismo de excreción del Fe mitocondrial (18). Hasta el momento solo se conocen dos proteínas implicadas en este proceso:
La frataxina es una proteína pequeña, codificada por un gen localizado en 9q13, que se encuentra deficiente en los pacientes con ataxia de Friedreich. El daño neuronal y cardiaco de esta enfermedad parece estar causado por el acúmulo de Fe en la mitocondria, por lo que se piensa que la frataxina tiene una función en la excreción mitocondrial de Fe (19).
En la anemia sideroblástica con ataxia ligada al cromosoma X (XLSA/A), se encuentran mutaciones de la proteína ABC 7 (ATP-Binding Cassette 7), relacionada con el transporte del grupo Hemo. Las alteraciones eritroides y neuronales de estos pacientes parecen tener su origen en el acúmulo de Fe mitocondrial (20).
ALTERACIONES MOLECULARES DE LA HEMOCROMATOSIS
Se considera en primer lugar la HC clásica en la que se puede demostrar mutación del gen HFE. Posteriormente se describen las sobrecargas férricas atípicas en las que se encuentran mutaciones de otros genes. En las bases de datos OMIM y Genbank se pueden encontrar los datos actualizados de las enfermedades genéticas y las mutaciones de sus genes. En la tabla I se resumen los datos de las alteraciones moleculares encontradas en las proteínas involucradas en el metabolismo del hierro.
GEN HFE
El descubrimiento y clonaje del gen de la HC marcó un hito en la identificación de genes asociados a enfermedades humanas. El mayor avance se produjo en 1977, cuando se encontró una elevada frecuencia del antígeno HLA A3 en pacientes no relacionados entre sí afectos de HC (1). La asociación fue confirmada en otras poblaciones, de manera que se pudo concluir que la HC era una enfermedad hereditaria, con una fallo genético localizado muy próximo al complejo mayor de histocompatibilidad de clase I (CPH I), en el brazo corto del cromosoma 6, y que se heredaba de forma autosómico recesiva (21-23). En 1996 se identificó el gen HFE o HLA-H mediante clonaje posicional, localizándose a 5 Mb teloméricos al HLA-A (2).
MUTACIÓN C282Y
La pérdida de función de la proteína HFE está causada en la mayoría de los pacientes con HC por una única y conservada mutación puntual en el aminoácido 282 (nucleótido 845 del exón 5) que convierte una cisteína en una tirosina (845 G → A) (2). Esta mutación C282Y (también denominada 845A) rompe un puente disulfuro en la hélice α3, eliminando su capacidad de unión a la β2-microglobulina, y evitando la expresión en la superficie de la célula de la proteína HFE (24). La introducción del gen mutado C260Y (equivalente al humano C282Y) en sistemas modelo (ratones knock-in) ha demostrado que la mutación no tiene un efecto de anulación completa del alelo, ya que se produce una sobrecarga férrica menor que en ratones sin el gen HFE. Además la sobrecarga no está presente en el momento del nacimiento, indicando que esta mutación provoca un aumento de absorción del hierro a nivel intestinal y no afecta a la captación placentaria de Fe (25). Sin embargo, la colocalización de la proteína HFE y de TfR en la membrana placentaria, parecería indicar alguna función en la homeostasis materno-filial del hierro implicando una posible participación en algunas formas de sobrecarga férrica neonatal (7).
El estudio de la prevalencia de esta mutación en 42 poblaciones de distintos países, ha determinado que el alelo C282Y tiene su origen en las poblaciones de origen céltico y nórdico. En el Reino Unido e Irlanda el 10-20% de la población es portadora, bajando hasta un 2-4% en los países del Sur y Este europeos (26).
MUTACIÓN H63D
Esta segunda mutación también denominada 187G, resulta de una cambio puntual en el nucleótido 187 (187 C → G) del exón 2 del gen HFE, originando la sustitución en el aminoácido 63 de una histidina por ác. aspártico (2). Las moléculas portadoras de la mutación H63D se procesan correctamente y se expresan en la membrana, uniéndose al TfR, pero al parecer no alteran la afinidad del receptor por la transferrina (27).
La mutación H63D tiene una distribución más global que C282Y, lo que indicaría un origen anterior. Las frecuencias alélicas más elevadas se localizan en Europa, Asia Menor y la India, siendo muy poco frecuente en África y América. España con un 25-30 % de portadores es uno de los países con mayor frecuencia de esta mutación.
Mutaciones minoritarias
Se han descrito otras mutaciones en el gen HFE que son mucho menos frecuentes y cuya importancia en la aparición del fenotipo de HC está aún por dilucidar.
--La delección de un nucleótido en el exón 3 (428delC) se descubrió en un paciente con HC que no era portador de ninguna de las mutaciones anteriores. Esta mutación causa un cambio en la pauta de lectura y un codón de parada prematuro (28).
--La mutación S65C, que resulta de la sustitución de una serina por una cisteína (nucleótido 193 A → T), está relacionada con una forma moderada de HC (29). Esta mutación se había descrito previamente como una simple variante que no conllevaba aparición de la enfermedad.
--Se han descrito cuatro nuevas mutaciones en un análisis realizado en población sudafricana de origen caucásico: los polimorfismos V53M (nucleótido 157 G → A) y V59M (nucleótido 175 G → A) en el exón 2; Q127H (nucleótido 381 A → C) en el exón 3; y R330M (nucleótido 989 G → T) en el exón 5. La mutación Q127H se localizó en un paciente con fenotipo de sobrecarga férrica severa portador de la mutación H63D del gen HFE y de la mutación R59W del gen PPOX (gen responsable de la porfiria variegata o porfiria genética sudafricana) (30).
--Se han identificado otras dos mutaciones en el exón 2, I105T (nucleótido 314 T → C) y G93R (nucleótido 277 G → C), en pacientes con HC y heterocigotos para la mutación H63D y C282Y respectivamente (31).
--Las mutaciones sin sentido en los codones 168 (G → T) y 169 (G → A) han sido descritas en pacientes italianos heterocigotos para la mutación C282Y y con HC (32).
--En los intrones 4 y 5 se han descrito cambios de nucleótidos: 5569 T → C y 5636 G → A y 5807 A → G y 6700 G → A, y mutaciones que originan un nuevo sitio de splicing (IVS3+1 G → T) (31,33).
--Por último, se ha encontrado que el gen puede ser escindido de formas alternativas (splicing alternativo) lo que genera varios tránscritos de los que al menos dos pueden tener actividad biológica (34).
HEMOCROMATOSIS NO LIGADAS A HFE
Hay un porcentaje relativamente elevado (15-40% según poblaciones) de individuos con sobrecarga férrica, que al parecer no son portadores de las mutaciones hasta ahora descubiertas en el gen HFE. Estos datos sugieren que puede haber otras mutaciones presentes en regiones no analizadas del gen (como la región del promotor), o una heterogeneidad genética, como en el caso de la HC juvenil.
Hemocromatosis juvenil o tipo II
La HC juvenil es una enfermedad autosómica recesiva, que cursa con sobrecarga férrica, clínica y genéticamente distinta de la HC. Aunque el patrón de los depósitos de hierro y los órganos afectados son similares, la HC juvenil manifiesta sus síntomas antes de los 30 años y afecta por igual a ambos sexos. Su clínica es más severa y cursa con disfunciones endocrinas, arritmias y fallos cardíacos. Si no es tratada a tiempo, la enfermedad es letal por las complicaciones cardíacas que origina.
Los pacientes con HC juvenil no son portadores de mutaciones en el gen HFE, ni ésta se encuentra ligada al cromosoma 6p. Se ha identificado el locus de la HC juvenil en el brazo largo del cromosoma 1 y se está intentado encontrar el gen responsable mediante clonaje posicional (35). Dado que en esta región del cromosoma 1 no se conoce ningún gen relacionado con la sobrecarga férrica, su identificación abrirá nuevas perspectivas para el estudio del metabolismo del hierro.
Hemocromatosis tipo III
Recientemente se ha encontrado un gen homólogo al del TfR localizado en 7q22 y denominado TFR2, del que se han identificado dos transcritos: TfR 2-α y TfR 2-β (8,36) Se ha demostrado que la expresión de TfR 2-α en el hígado no se ve inhibida por la acumulación de hierro en los hepatocitos, como ocurre con el TfR clásico, lo que explicaría la susceptibilidad del hígado a la sobrecarga férrica en las HC (8,36). Alteraciones de estos genes se han encontrado en dos familias sicilianas diagnosticadas de HC no vinculada al gen HFE, y portadoras de una mutación Y250X en el gen TFR2, que provoca un codón de parada prematura (37).
Hemocromatosis autosómica dominante (Islas Salomón)
En 1990 se describió el caso de una extensa familia de origen melanesio con una forma autosómica dominante de HC hereditaria, sin que se conozca si tiene relación con el gen HFE (39).
OTRAS SOBRECARGAS FÉRRICAS
Sobrecarga férrica neonatal
Aproximadamente se han descrito en la literatura unos 100 casos de esta enfermedad. Aunque el exceso de depósito de hierro con el consiguiente daño hepático podría ser consecuencia de procesos muy diversos, como tirosinemia hereditaria o infecciones virales maternas, parece que ciertos subtipos tienen un carácter hereditario. Actualmente se está trabajando en el mapeo sistemático para la localización del locus del gen responsable.
Sobrecarga férrica africana
A diferencia con la HC, que en la población de origen caucásico es de carácter hereditario, la sobrecarga férrica en las poblaciones sub-saharianas tradicionalmente se ha atribuido al consumo de cerveza, elaborada artesanalmente en recipientes de hierro. Sin embargo los últimos estudios realizados parecen indicar un carácter genético que no estaría vinculado ni al gen HFE ni al locus. Al parecer la heterocigosidad para este gen desconocido causa susceptibilidad a la sobrecarga de Fe, mientras que los homocigotos están considerablemente afectados (38). El patrón de los depósitos de hierro es distinto al de la HC, acumulándose principalmente en las células de Kupfer y en los hepatocitos, originando cirrosis y carcinoma hepatocelular, siendo poco frecuente la diabetes o las cardiomiopatías. Una importante consecuencia clínica a destacar es la conveniencia de incluir a los individuos con ancestros africanos en los programas de screening de sobrecarga férrica.
Atransferrinemia
La Atransferrinemia es una enfermedad autosómica recesiva poco frecuente en la que no hay transferrina plasmática o su concentración es muy baja (hipotransferrinemia). Sólo se han descrito ocho casos hasta la fecha. Desde el nacimiento hay una anemia hipocrómica microcítica severa, que sólo responde a la infusión de transferrina o a las transfusiones de sangre. Los individuos afectados pueden desarrollar sobrecarga férrica, ya que el hierro que se absorbe de la dieta circula unido a otros constituyentes sanguíneos y se deposita principalmente en el hígado, páncreas, corazón, tiroides y riñones, mediante mecanismos independientes de la transferrina. Sin embargo, este hierro prácticamente no se puede utilizar para la producción de eritrocitos al no haber mecanismos alternativos para su entrada en los precursores eritroides (40).
Aceruloplasminemia
La aceruloplasminemia es una enfermedad poco frecuente del metabolismo del hierro, que aparece de forma autosómica recesiva, producida por la falta de actividad ceruloplasmin ferroxidasa, debido a mutaciones en el gen de la ceruloplasmina (15). Los individuos afectados padecen diabetes mellitus y una neurodegeneración progresiva de la retina y de los ganglios basales en la edad adulta. Se produce un acusado depósito de hierro en el hígado, páncreas y cerebro, y acúmulos menores en el bazo, corazón, riñón, tiroides y retina. Los niveles de cobre y hierro sérico son bajos, la ferritina sérica moderadamente elevada y no se detecta ceruloplasmina.
Se han descrito diversas mutaciones en las familias afectas localizadas en el gen de la ceruloplasmina que se ubicado en el cromosoma 3q (41). La afectación del sistema nerviosos central por la acumulación de hierro, diferencia a la aceruloplasminemia del resto de enfermedades que cursan también con sobrecarga. Al parecer en determinadas células gliales la expresión del gen de la ceruloplasmina es esencial para la homeostasis del hierro y la supervivencia neuronal en el sistema nervioso central.
El tratamiento de los pacientes con agentes quelantes del hierro, como la deferoxamina, disminuye los depósitos de hierro en el cerebro y detiene la progresión de los síntomas neurológicos. Por este motivo, la detección precoz de la aceruloplasminemia hereditaria es fundamental para evitar el deterioro neurológico (42).
Hiperferritinemia con catarata congénita autosómica dominante
La hiperferritinemia con catarata congénita autosómica dominante, es una enfermedad del metabolismo del hierro que se caracteriza por catarata bilateral precoz y concentraciones elevadas de ferritina sérica (14). La ferritina se encuentra formada por 24 subunidades de al menos dos tipos L y H. En individuos normales hay una mezcla de ambos tipos, mientras que en los afectados por esta enfermedad la subunidad H es indetectable. Aparte de la aparición de cataratas, no hay otras manifestaciones clínicas ni alteraciones hematológicas o bioquímicas.
Los estudios moleculares han identificado múltiples mutaciones puntuales en el elemento IRE (elemento de respuesta al hierro) del RNA mensajero de la L-ferritina, afectando al motivo CAGUGU que forma parte del bucle IRE y que interviene en la unión de IRP (proteína reguladora del hierro). Estas mutaciones impedirían la unión de IRP provocando una síntesis constitutiva y sin regulación de la L-ferritina (43).
Ataxia de Friedreich
La ataxia de Friedrich es la ataxia espinocerebelosa hereditaria más frecuente, tiene un patrón de aparición autosómico recesivo y se caracteriza por una degeneración progresiva que afecta al sistema nervioso central, periférico, y corazón. El origen de la enfermedad se encuentra en la carencia de frataxina, una proteína mitocondrial que regula la expulsión de hierro en la mitocondria. La acumulación de hierro mitocondrial provoca un estrés oxidativo y fallos en la cadena respiratoria que causan daño celular (44).
Anemia sideroblástica ligada al cromosoma X
Aunque existen casos sin aparente afectación familiar previa, la mayoría de las anemias sideroblásticas tienen un patrón hereditario. Las más frecuentes presentan herencia ligada al sexo y afectan fundamentalmente a los varones, aunque las mujeres portadoras puedan excepcionalmente sufrir anemia leve o rasgos hematológicos característicos de la enfermedad. Se han descrito pocos casos bien documentados de anemia sideroblástica constitucional de herencia autosómica, dominante o recesiva. El trastorno enzimático mejor caracterizado en estas anemias congénitas es el déficit de ALA-sintetasa (delta-aminolevulinato sintetasa), sobre todo en las formas hereditarias ligadas al cromosoma X. Este déficit se debe a mutaciones puntuales del gen ALAS2 localizado en Xp11.21 (45). Los individuos afectados desarrollan anemia en la infancia y muerte por sobrecarga férrica al cabo de pocos años.
Otra forma de anemia sideroblástica hereditaria es la ligada al cromosoma X con ataxia (XLSA/A). Se caracteriza por una ataxia espinocerebelosa no progresiva que se manifiesta en los primeros años de vida. La mutación responsable afecta a un gen localizado en Xq13 que codifica la proteína ABC7 (ATP-binding cassette 7), que está implicada en el transporte del grupo hemo desde la mitocondria. Las mutaciones en ABC7 producen una acumulación de hierro en la mitocondria que afecta principalmente a las células neuronales y eritroideas (46).
Bibliografía
1. Simon M, Bourel M, Genetet B. and Fauchet R. Idiophatic hemochromatosis: demonstration of recessive transmission and early detection by family HLA typing. N Engl J Med 1977; 297: 1017-1021. [ Links ]
2. Feder JN, Gnirke A, Thomas W, et al. A novel MHC class-I like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996; 13: 399-408. [ Links ]
3. Andrews NC, Fleming MD, Levy JE, Molecular insights into mechanisms of iron transport. Curr Opin Hematol 1999; 6: 61-64 [ Links ]
4. Vulpe CD, Kuo YM, Murphy TL. Hephaestine, a ceruloplasmin homologue implicated in intestinal iron. transport, is defective in the sla mouse. Nat. Genet. 1999; 21: 195-199. [ Links ]
5. Ponka P, Beaumont C, Richardson DR, Function and regulation of transferrin and ferritin. Semin Hematol 1998; 35: 35-54. [ Links ]
6. Parkkila S, Waheed A, Britton RS, et al. Association of the transferrin receptor in human placenta with HFE, the protein defective in hemochromatosis. Proc Natl Acad Sci USA 1997; 94: 13198-13202. [ Links ]
7. Kawabata H, Yang R, Hirama T. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem 1997; 274: 20826-20832. [ Links ]
8. Andrews NC. Iron homeostasis: Insights from genetics and animal models. Nat Rev Genet 2000; 1: 208-217 [ Links ]
9. Bennettt MJ, Lebron JA, Bjorkman PJ. Cristal structure of the hereditary haemocromatosis protein HFE complexed with the transferrin receptor. Nature 2000; 403: 46-53. [ Links ]
10. Zhou XY, Tomatsu S, Fleming RE, et al. HFE gene knockout produces mouse model of hereditary hemochromatosis. Proc Natl Acad Sci 1998; 95: 2492-2497. [ Links ]
11. Levy JE, Montross LK, Andrews NC. Genes that modify the HC phenotype in mice. J Clin Invest 2000; 105: 1209-1217. [ Links ]
12. Yu J, Wessling-Resnick M. Expression of SFT is enhanced by iron chelation in HeLa cells and by hemochromatosis in liver. J Biol Chem 1998; 273: 34675-34678. [ Links ]
13. Sheth S, Brittenham GM. Genetic disorders affecting proteins of iron metabolism: Clinical implications. Ann Rev Med 2000; 51: 443-464. [ Links ]
14. Bonneau D, Winter-Fuseau I, Loiseau MN, et al. Bilateral cataract and high serum ferritin: a new dominant genetic disorder? J Med Genet 1995; 32: 778-779. [ Links ]
15. Gitlin JD. Aceruloplasminemia. Pediatr Res 1998; 44: 271-276. [ Links ]
16. Rouault T, Klausner R. Regulation of iron metabolism in eukaryotes. Curr Top Cell Regul, 1997; 35: 1-19 [ Links ]
17. Rouault TA, Klusner RD. Iron sulfur clusters as biosensors of oxidants and iron. Trends Biochem Sci 1997; 21: 174-77. [ Links ]
18. Radiski DC, Babcock MC, Kaplan J. The yeast Frataxin Mitochondrial Iron Efflux: Evidence for a mitochondrial iron cycle. J Biol Chem 1999; 274: 8, 4497-4499. [ Links ]
19. Knigth SA, Kim R, Pain D, Dancis A. The yeast connection to Friedreich ataxia. Am J Hum Genet 1999; 64: 365-371. [ Links ]
20. Allikmets R, Raskind WH, Hutchinson A. Mutations of a putative mitochondrial iron transporter gen (ABC 7) in XLSA/A. Human Mol Genet 1999; 8: 743-749. [ Links ]
21. Dorán T, Bashir HV, Trejaut J, et al. Idiopathic hemochromatosis in the Australian population: HLA linkage and recessivity. Human Immunol 1981; 2: 191-200. [ Links ]
22. Santos M, Schilham MW and Rademakers LPHM, et al. Defective iron homeostasis in b2-microglobulin knockout mice recapitulates hereditary hemochromatosis in man. J Exp Med 1996; 184: 1975-1985. [ Links ]
23. Rothenberg BE, Voland JR. Beta-2-microglobulin knockout mice develop parenchymal iron overload: a putative role for class I genes of the major histocompatibility complex in iron metabolism. Proc Natl Acad Sci USA 1996; 93: 1529-1534. [ Links ]
24. Feder JN, Tsuchihashi Z, Irrinki A, et al. The hemochromatosis founder mutation in HLA-H disrupts beta2-microglobulin interaction and cell surface expression. J Biol Chem 1997; 272: 14025-14028. [ Links ]
25. Levy JE, Montross L.K, Cohen DE, et al. The C282Y mutation causing hereditary hemochromatosis does not produce a null allele. Blood 1999; 94: 9-11. [ Links ]
26. Merryweather-Clarke AT, Pointon JJ, Shearman JD, Robson KJ. Global prevalence of putative haemochromatosis mutations. J Med Genet 1997; 34: 275-278. [ Links ]
27. Feder JN, Penny DM, Irrinki A, et al. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc Natl Acad Sci USA 1998; 95: 1472-1477. [ Links ]
28. Pointon JJ, Shearman JD, Merryweather-Clarke AT, Robson KJH. A single nucleotide deletion in the putative haemochromatosis gene in a patient who is negative for the C282Y and H63D mutations. In: Iron in Biology and Medicine, p. 268. VIth Conference of the International Association for the Study of Disorders in Iron Metabolism and the XIIIth International Conference on Proteins of Iron Metabolism. St. Malo, France, 1997; 16-20. [ Links ]
29. Mura C, Raguenes O, Ferec C. HFE mutations analysis in 711 hemochromatosis probands: evidence for S65C implications in mild form of hemochromatosis. Blood 1999; 93: 2502-2505. [ Links ]
30. De Villiers JN, Hillermann R, Louser L, Kotze MJ. Spectrum of mutations in the HFE gene implicated in haemochromatosis and porphyria. Hum Mol Genet 1999; 8: 1517-1522. [ Links ]
31. Barton JC, Sawada-Hirai R, Rothenberg BE, Acton RT. Two novel missense mutations of HFE gene I105T and G93R and identification of the S65C mutation in Alabama hemochromatosis probands. Blood Cells Mol Dis 1999; 25: 147-155. [ Links ]
32. Piperno A, Arosio C, Fossati L, et al. Two novel nonsense mutations of HFE gene in five unrelated patients with hemochromatosis. Gastroenterology 2000; 119: 441-445. [ Links ]
33. Wallace DF, Dooley JS, Walker AP. A novel mutation of HFE explains the classical phenotype of genetic hemochromatosis in a C282Y heterozygote. Gastroenterology 1999; 116: 1409-1412. [ Links ]
34. Thenie A, Orhant M, Gicquel I, et al. The HFE gene undergoes alternate splicing processes. Blood Cells Mol Dis 2000; 26: 155-162. [ Links ]
35. Roetto A, Totaro A, Cazzola M, et al. Juvenile hemochromatosis locus maps to chromosome 1q. Am J Hum Genet 1999; 64: 1388-1393. [ Links ]
36. Fleming RE, Migas MC, Holden CC, et al. Transferrin receptor 2: continued expression in mouse liver in the face of iron overload and in hereditary hemochromatosis. Proc Natl Acad Sci USA 2000; 97: 2214-2219. [ Links ]
37. Camaschella C, Roetto A, Cali A, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nature Genet 2000; 25: 14-15. [ Links ]
38. Moyo VM, Mandishona E, Hasstedt SJ, et al. Evidence of genetic transmission in african iron overload. Blood 1998; 91: 1076-1082. [ Links ]
39. Eason RJ, Adams PC, Aston CE, Searle J. Familial iron overload with possible autosomal dominant inheritance. Aust NZ J Med 1990; 20: 226-230. [ Links ]
40. Levy JE, Jin O, Fujiwara Y, et al. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat Genet 1999; 21: 396-399. [ Links ]
41. Yazaki M, Yoshida K, Nakamura A, et al. A novel splicing mutation in the ceruloplasmin gene responsible for hereditary ceruloplasmin deficiency with hemosiderosis. J Neurol Sci 1998; 156: 30-34. [ Links ]
42. Miyajima H, Takahashi Y, Kamata T, et al. Use of desferroxamine in the treatment of aceruloplasminemia. Ann Neurol 1997; 41: 404-407. [ Links ]
43. Cazzola M, Bergamaschi G, Tonon L, et al. Hereditary hyperferritinemia-cataract syndrome: relationship between phenotypes and specific mutations in the iron-responsive element of ferritin light-chain mRNA. Blood 1997; 90: 814-821. [ Links ]
44. Radisky DC, Babcock MC, Kaplan J. The yeast frataxin homologue mediates mitochondrial iron efflux. Evidence for a mitochondrial iron cycle. J Biol Chem 1999; 274: 4497-4499. [ Links ]
45. Cotter PD, Willard HF, Gorski JL, Bishop DF. Assignement of human erythroid delta-aminolevulinate synthase ALAS2 to a distal subregion of band Xp11.21 by PCR analysis of somatic cell hybrids containing X:autosome translocations. Genomics 1992; 13: 211-212. [ Links ]
46. Shimada Y, Okuno S, Kawai A, et al. Cloning and chromosomal mapping of a novel ABC transporter gene (hABC7) , a candidate for X-linked sideroblastic anemia with spinocerebellar ataxia. J Hum Genet 1998; 43: 115-122. [ Links ]
BASES DE DATOS ELECTRÓNICAS
Online Mendelian Inheritance in Man (OMIM),
http://www.ncbi.nlm.nih.gov/Omim/
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/index.html

