










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versión impresa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.78 no.7 jul. 2003
REVISIÓN
VÍAS DE SEÑALIZACIÓN INTRACELULAR QUE CONDUCEN
A LA APOPTOSIS DE LAS CÉLULAS DE LA RETINA
INTRACELLULAR PATHWAYS LEADING TO APOPTOSIS OF
RETINAL CELLS
GARCÍA M1, VECINO E1
| RESUMEN Algunas patologías oculares como la retinitis pigmentosa, la degeneración macular asociada a la edad, el glaucoma, el desprendimiento de retina, la retinopatía diabética o la miopía patológica producen la muerte por apoptosis de las células de la retina. Las caspasas son proteasas que participan en la cascada de señalización intracelular que conduce a la apoptosis en diferentes tipos celulares. Utilizando distintos modelos experimentales se ha demostrado la activación de caspasas en las células de la retina tras un daño. Sin embargo, algunos trabajos muestran que otras moléculas además de las caspasas se activan en la retina durante la apoptosis. Además el tipo de caspasa que se activa parece ser diferente dependiendo del tipo de célula de la retina que se analice y dependiendo del tipo de daño. Palabras clave: Apoptosis, caspasa, retina, neuroprotección, mitocondria.
| SUMMARY Some ocular diseases, such as retinitis pigmentosa, age-related macular degeneration, glaucoma, retinal detachment, diabetic retinopathy or pathological myopia result in apoptotic death of retinal cells. Caspases are proteases that take part in the intracellular signaling pathways that cause apoptosis in different cell types. By using different experimental models, an increase in caspase activity in retinal cells after damage has been shown. However, some studies have shown that other molecules apart from caspases are activated during apoptosis in the retina. Moreover, the type of caspase that is activated seems to be different depending on the type of retinal cell and the pathology analysed. Key words: Apoptosis, caspase, retina, neuroprotection, mitochondria.
|
Recibido:30/6/03. Aceptado: 14/7/03.
Departamento Biología Celular. Facultad de Medicina. Universidad del País Vasco. Lejona. Vizcaya. España.
1 Doctora en Biología.
Proyecto subvencionado: Comunidad Europea (Pro Age Ret QLK6-2000-00569) y Universidad del País Vasco (1/UPV00075.327-E-14887/2002).
Los autores manifiestan que no tienen interés comercial ni han recibido apoyo económico.
Correspondencia:
Elena Vecino
Dpto. Biología Celular e Histología
Facultad de Medicina. Universidad del País Vasco
48940 Leioa (Vizcaya)
España
E-mail: gcpvecoe@lg.ehu.es
INTRODUCCIÓN
Frente a la muerte accidental de las células debida a una lesión aguda, la muerte celular programada es un proceso activo que se caracteriza por una alteración morfológica denominada apoptosis. Los cambios morfológicos observados durante la apoptosis incluyen: fragmentación del ADN cromosómico, condensación de la cromatina, acidificación del citoplasma y ruptura del núcleo. Finalmente, la célula se encoge y se rompe en fragmentos rodeados por membrana denominados cuerpos apoptóticos (1), visibles claramente a microscopia óptica (fig. 1). Las células apoptóticas son reconocidas y fagocitadas por macrófagos o células adyacentes, y son retiradas de los tejidos de forma eficaz (1,2).
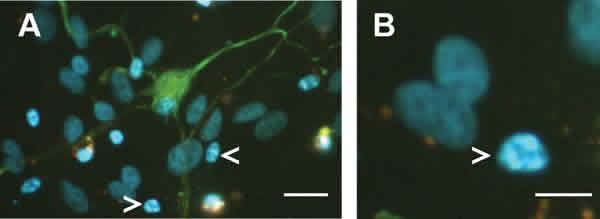
Fig. 1. Células ganglionares de la retina de cerdo tras 7 días de cultivo sobre sustrato de laminina-polilisina
en medio de cultivo químicamente definido. Los núcleos se identificaron utilizando 4,6-diaminodiphenyl-2-
phenylindole (DAPI) y las células se observaron con un microscopio de fluorescencia. Aunque la mayoría de los
núcleos presentan una morfología normal, se pueden apreciar algunos núcleos fragmentados correspondientes
a células apoptóticas (flechas). Escala A: 25 µm; escala B: 5 µm.
La muerte celular programada es un mecanismo fisiológico fundamental durante el desarrollo embrionario y para el mantenimiento de los tejidos adultos.
Durante el desarrollo, el proceso de apoptosis desempeña un papel clave eliminando las células innecesarias en diversidad de tejidos. Un caso bien caracterizado de muerte celular programada lo proporciona el desarrollo del sistema nervioso de los mamíferos. En este proceso, se produce un número excesivo de neuronas que son eliminadas en parte durante el desarrollo. Así, en el caso de la retina de rata (fig. 2) se estima que el 90% de las células ganglionares mueren durante la primera semana postnatal (3). Aquellas que sobreviven, se seleccionan tras realizar correctamente la conexión con sus células diana, las cuales secretan factores de crecimiento que permiten la supervivencia celular mediante el bloqueo de la vía de señalización que conduce a la apoptosis.
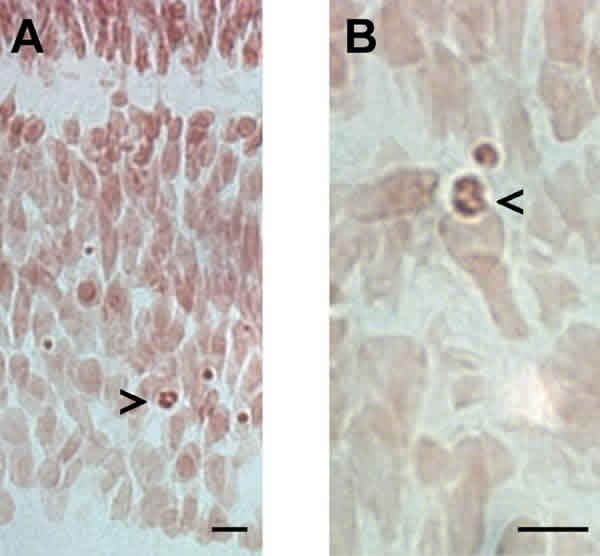
Fig. 2. Secciones de retina de rata postnatal (9 días) teñidas mediante la técnica de Feulgen para observar las
células en apoptosis. En la figura A se muestra una célula en la capa nuclear interna que presenta el núcleo
claramente apoptótico. En la figura B se muestra otra célula de la misma capa a mayor aumento. Escala 10 µm.
En el adulto, la muerte celular programada es la responsable de mantener constante el número de células en tejidos sometidos a un continuo recambio celular. Además, la muerte celular programada constituye un mecanismo de defensa mediante el cual las células alteradas, potencialmente peligrosas, son eliminadas del organismo. El fallo del mecanismo de apoptosis se ha relacionado con el desarrollo de enfermedades neurodegenerativas, autoinmunes y algunos tipos de cáncer (4).
La muerte por apoptosis puede desencadenarse por diferentes señales intra o extracelulares. La naturaleza de los inductores es diversa y un mismo estímulo puede generar efectos diferentes y hasta opuestos en distintos tipos celulares, e incluso en células del mismo tipo que se encuentran en distinta etapa de desarrollo (4).
Apoptosis y caspasas
Las caspasas son proteasas que juegan un papel clave en el proceso de apoptosis (5). La familia de las caspasas está constituida por más de doce proteasas caracterizadas por la presencia de residuos de cisteína en su sitio activo. Estas proteínas están ampliamente conservadas en el proceso evolutivo y comparten secuencias estructurales comunes (6). Las caspasas se sintetizan como precursores inactivos que se convierten en la forma activa por rotura proteolítica. Una vez activas, las caspasas producen la hidrólisis a partir de residuos de ácido aspártico en la proteína sustrato. Así, la activación inicial de una caspasa provoca una reacción en cadena que conduce a la activación de otras caspasas y a la muerte de la célula. Por lo tanto, la regulación de la activación de las caspasas es fundamental para determinar la supervivencia celular (5).
Aunque la familia de las caspasas comparte una estructura común, los análisis filogenéticos han demostrado la existencia de subfamilias que presentan diferentes características y que por lo tanto pueden tener distinto papel en el proceso de apoptosis (6,7). Así, las caspasas iniciadoras (caspasas -2, -8, -9, -10) presentan pro-dominios funcionales que permiten la asociación de estas moléculas con complejos de señalización; las caspasas ejecutoras (caspasas -3, -6 ,-7) son activadas por las caspasas iniciadoras y son las responsables de la interacción con otras moléculas que desencadenan la apoptosis. La tercera subfamilia de caspasas es la de las caspasas procesadoras de citoquinas (caspasas-1, -4, -5, -12, -13, -14).
Se ha descrito que las caspasa 1 se activa por la ausencia de factores de crecimiento (8), que hidroliza a la caspasa 3 in vitro y que promueve el procesamiento y activación de la interleucina-1ß (IL-1ß), sustancia implicada en la muerte neuronal (9). En cuanto a la caspasa 2, se sabe que se activa tras su unión a una molécula adaptadora que a su vez se une a una parte del complejo de señalización del receptor de factor de necrosis tumoral (TNF). La caspasa 2 activa puede procesar su propio precursor (10).
La caspasa 9, que en situación fisiológica se encuentra en su forma inactiva en el citosol, se activa tras la salida de citocromo c desde la mitocondria. Ante un determinado daño se produce una alteración en la membrana mitocondrial que desencadena la salida de citocromo c al citosol. El citocromo c forma entonces un complejo con el factor activador de proteasa (Apaf-1), dATP y procaspasa 9, lo que conduce a la activación de la caspasa. Una vez activa, la caspasa 9 puede activar otras caspasas (11) (fig. 3).
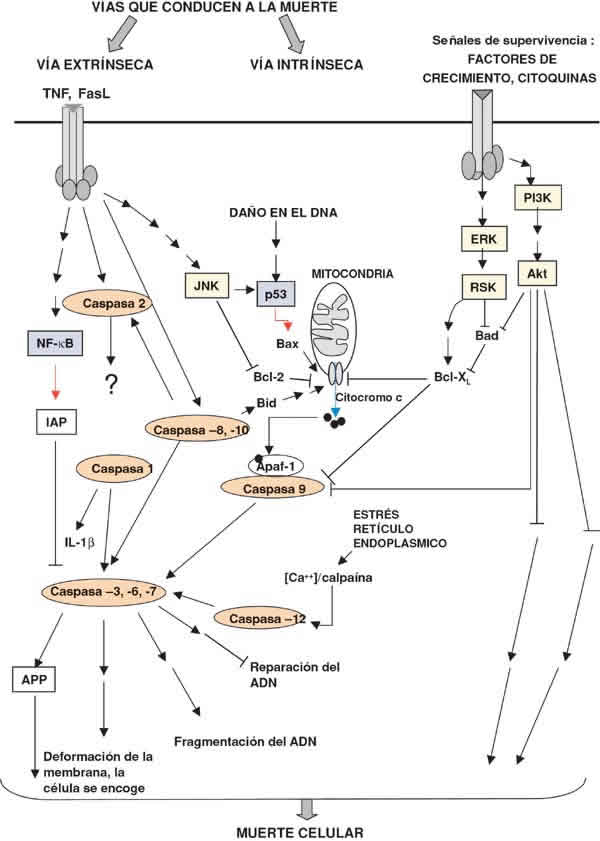
Fig. 3. Esquema de las principales vías de señalización que conducen a la apoptosis o supervivencia de las
células. Los estímulos que conducen a la apoptosis de la célula pueden ser intracelulares (vía intrínseca) o
extracelulares (vía extrínseca). Un paso clave en la vía intrínseca es la salida de citocromo c desde la mitocondrial
y, por lo tanto la salida de citocromo c desde la mitocondria está regulada por miembros de la familia Bcl-2,
alguno de estos miembros son pro-apoptóticos como Bid y Bax mientras que otros son antiapoptóticos (Bcl-XL y
Bcl-2). Otro mecanismo de apoptosis desencadenada por estímulos intracelulares se produce por estrés del
retículo endoplásmico. Este proceso está mediado por caspasa 12. La vía extrínseca se activa tras la unión de un
ligando, polipéptido de la familia del TNF, a su receptor esto activa a las caspasas iniciadoras (2, 8, 10). Una vez
activadas, estas caspasas escinden y activan otras caspasas efectoras (caspasas 3, 6 y 7) que en última instancia
son las responsables de la escisión de proteínas del cito-esqueleto y proteínas nucleares, lo que conduce a la
apoptosis de las células. Los ligandos antiapoptótico, entre los que se incluyen los factores de crecimiento y las
citoquinas activan Akt y RSK, inhibiendo Bad y evitando la salida de citocromo c. El receptor de TNF puede inhibir
la apoptosis mediante una vía mediada por NF-kB que incluye la inducción del inhibidor de apoptosis (IAP) que
inhibe directamente las caspasas 3, 7 y 9. Abreviaturas: Apaf-1: Factor activador de la proteasa apoptótica. APP:
Proteína precursora ß-amiloide. Ask: Quinasa reguladora de la señal de apoptosis. Bcl-2: proto-oncogen del linfoma
de los linfocitos B, ErK: Quinasa regulada por señales extracelulares. IAP: Inhibidor de apoptosis. JNK: Jun quinasa
N-terminal. RSK: quinasa ribosómica S6 90 kDa. TNF: Factor de necrosis tumoral. NF-kB: Factor nuclear kappa
B. P13K: Fosfoinositido 3 quinasa. En amarillo se muestran las moléculas que actúan como quinasas y en azul los
factores de transcripción. Las flechas sencillas muestran las acciones directas mientras que las flechas dobles
muestran un efecto en varios pasos. Las flechas acabadas en punta: efecto activador, las terminadas con una
línea: efecto inhibidor. La flecha azul indica translocación y la roja estimulación de la transcripción.
Las caspasas 3, 6 y 7 son las principales responsables de los cambios morfológicos y bioquímicos que ocurren en las células apoptóticas. Entre sus sustratos se encuentran un factor responsable de la fragmentación del ADN (12). Además las caspasas eliminan la poli (ADP-ribosa) polimerasa (PARP) (13), molécula implicada en la maquinaria celular que repara el daño en el ADN. Por otro lado, activan la vía que conduce a la condensación de la cromatina (12) y participan en la destrucción de la lámina nuclear (14) y de las proteínas del citoesqueleto (5).
Vía intrínseca de la apoptosis
La vía mitocondrial se activa por estrés y otras señales que provocan la salida de citocromo c desde la mitocondria (15).
Bajo condiciones normales de supervivencia celular, el citocromo c se localiza en el espacio intermembrana mitocondrial; sin embargo, algunos estímulos provocan la liberación de citocromo c al citosol, lo que se acompaña de pérdida del potencial de membrana y desestabilización de la membrana externa de la mitocondria. En el citosol, el citocromo c se une a Apaf-1 y una vez unido recluta y activa a la procaspasa 9, la cual puede a su vez activar otras caspasas (11).
En mamíferos se ha descrito la presencia de una familia de proteínas, familia Bcl-2 (proto-oncogen del linfoma de los linfocitos B), implicada en el control de la apoptosis mediante el control de la permeabilidad mitocondrial y la liberación de citocromo c (16). Algunos miembros de la familia Bcl-2, como el propio Bcl-2 y Bcl-XL se encuentran en la membrana externa de la mitocondria e inhíben la liberación de citocromo c, son por lo tanto anti-apoptóticos. El efecto anti-apoptótico de Bcl-2 implica el bloqueo de Bax y la inhibición de la liberación de citocromo c desde la mitocondria (17). Algunos autores han sugerido que Bcl-XL puede inhibir la apoptosis mediante la interacción con Apaf-1, lo que bloquea la formación del complejo Apaf-1/procaspasa 9 (18).
Otros miembros de la familia Bcl-2 como Bcl-XS, Bad, Bid y Bax actúan promoviendo la liberación de citocromo c desde la mitocondria. Estos miembros que inducen el daño en la mitocondria residen en el citosol y se translocan a la mitocondria ante un estimulo (fig. 3).
La apoptosis puede también producirse por estrés del retículo endoplásmico. Se ha demostrado que el estrés del retículo, producido por alteración en la homeostasis de calcio o la acumulación de proteínas, conduce a la activación de la caspasa 12 (19) (fig. 3).
Vía extrínseca de la apoptosis
Algunos polipéptidos, que pertenecen a la familia del factor de necrosis tumoral (TNF), señalizan la muerte celular programada a través de la activación de receptores. La unión de uno de esos ligandos a su receptor genera la señal de apoptosis activando directamente las caspasas (20). La vía extrínseca de apoptosis comienza cuando el TNF y otros miembros similares como FasL se unen al receptor produciendo una trimerización del receptor y el reclutamiento de un complejo que contiene proteínas adaptadoras. Estas proteínas adaptadoras permiten la unión de la procaspasa 8 favoreciendo su autoactivación (fig. 3).
Una vez activa, la caspasa 8 puede activar a otras caspasas posteriores (16) iniciando de esta manera una cascada de caspasas que conduce a la muerte celular.
Además, la caspasa-8 puede también activar a un miembro pro-apoptótico de la familia Bcl-2 denominado Bid, Bid es uno de los miembros de la familia Bcl-2 que induce la apoptosis (21). Normalmente se encuentra en forma inactiva en el citosol, sin embargo, la ruptura por la caspasa-8 permite a Bid translocarse a la mitocondria, donde altera la membrana y libera el citocromo c al citosol. Esto conduce a la activación de la caspasa-9 amplificando aun más la cascada de caspasas que se inició por la activación directa de la caspasa-8 mediante los receptores implicados en la muerte celular (fig. 3).
La vía del receptor TNF-R1 puede, aparte de conducir a la apoptosis, activar una vía de señalización, dependiente del factor nuclear kappa-B (NF-kappa-B), que conduce a la supervivencia de la célula. El NF-kappa-B es un factor que en situación normal se encuentra en el citoplasma de las células en estado inactivo debido a su unión con la proteína inhibidora Ikappa-B. La activación del receptor de TNF produce la fosforilación y degradación de Ikappa-B liberándose el NF-kappa-B que se transloca al núcleo activando distintos genes entre los que se incluyen inhibidores de caspasas (fig. 3).
Vías de supervivencia celular
Algunas vías de señalización promueven la supervivencia celular mediante la inhibición de la apoptosis. Estas vías de señalización controlan el destino de una gran variedad de células cuya supervivencia depende de factores de crecimiento extracelulares o de interacciones célula-célula. Entre los primeros, figuran las neurotrofinas y en particular el factor de crecimiento derivado del cerebro (BDNF).
Una de las principales vías de señalización intracelular que promueve la supervivencia de la célula comienza en la enzima fosfoinositido-3 quinasa (PI3K), que es activada bien por la proteína tirosina quinasa o bien por receptores asociados a proteínas G. La PI3K activa indirectamente a la proteína serina/treonina quinasa Akt . Un sustrato de Akt es un miembro de la familia de Bcl-2 denominado Bad. Bad es uno de los miembros de la familia de Bcl-2 que, como Bid, induce la muerte celular estimulando la liberación de citocromo c desde la mitocondria. La fosforilación de Bad por Akt genera un sitio de unión para las proteínas que secuestran a Bad en el citosol, impidiendo de esta manera la translocación de Bad a la membrana mitocondrial (22). Akt también puede bloquear directamente la activación de las caspasas mediante la fosforilación de la caspasa 9.
Otra vía de señalización que conduce a la supervivencia celular es la vía de las Ras/Raf/MAPK (proteínas quinasas activadas por mitógeno). El mecanismo mediante el cual esta vía inhibe la apoptosis implica la fosforilación y activación por la quinasa regulada por señales extracelulares (ERK) de una proteína quinasa denominada quinasa ribosómica S6 90 kDa (RSK). Al igual que Akt, RSK fosforila a Bad (23), de tal manera que Bad es un punto de convergencia de las vías de la PI3K/Akt y de las MAPK en la señalización de la supervivencia celular. Además ERK y RSK pueden fosforilar factores de transcripción que afectan a la expresión de genes que regulan la apoptosis (fig. 3).
APOPTOSIS EN LA RETINA
Cuando se comparan los procesos que conducen a la apoptosis de células retinianas en cultivo tras un daño isquémico, excitotóxico, o tras la administración del anticuerpos contra la proteína de shock térmico, hsp27, se observa que tienen en común la activación de la cascada de proteolisis, incluyendo caspasa 3 y la escisión de PARP. Una excepción es que la activación de caspasa 8 no se observa durante la apoptosis inducida por anticuerpo hsp27. Además, en todos los casos, durante la apoptosis se observa un descenso en la expresión de Bcl-2 y un aumento de Bax (24). Otro trabajo que ha analizado los mecanismos de muerte de los distintos tipos de células de la retina ante distinto daño, ha demostrado un aumento del marcaje para la Jun quinasa N-terminal (JNK) en el citoplasma de células apoptóticas, independientemente del tipo celular que se analice, del grado de diferenciación de las células y del método de inducción de apoptosis (25).
Aunque estos trabajos muestran que existen mecanismos comunes que conducen a la apoptosis de las células cuando se produce un daño también se ha descrito que algunas moléculas se expresan durante la apoptosis sólo en algunos tipos celulares y en respuesta a determinados tipos de daño. A continuación describiremos las rutas intracelulares que parecen estar implicadas en la apoptosis de las células de la retina.
Apoptosis de los fotorreceptores
Algunos trabajos han puesto de manifiesto la activación de distintos tipos de caspasas en la cascada de señalización que conduce a la apoptosis de los fotorreceptores ante un daño. Así, tras inducir la degeneración de los fotorreceptores mediante la aplicación de ciclos de luz azul en ratas, se produce un incremento en la expresión de la forma inactiva de la caspasa 3, la procaspasa 3, además de un aumento de su activación (26). Esta caspasa está también implicada en la muerte de los fotorreceptores tras la inyección intraperitoneal de M-methyl-N-nitrosourea en ratas (27).
La caspasa 1 se activa en otro modelo experimental en ratones, en el que la degeneración de los fotorreceptores se induce mediante la aplicación de niveles altos de luz visible (28), además la expresión de los factores de transcripción asociados a apoptosis c-Jun y c-Fos aumenta temporalmente en este modelo de daño (28). Ambos factores son componentes del factor de transcripción AP-1, que activa la transcripción de varios genes diana en las células.
La caspasa 1 junto con la caspasa 2, también parece mediar la muerte por apoptosis de los fotorreceptores de ratas Royal College Surgeons, utilizadas como modelo para el estudio de retinitis pigmentosa. La activación de las caspasas en esta patología está mediada por calcio ya que la administración intraperitoneal del agonista de calcio nilvadipina previene la muerte de los fotorreceptores (29). La caspasa 1 está también implicada en la apoptosis tras la exposición a luz visible de fotorreceptores en cultivo procedentes de retina de ratón. Utilizando este modelo in vitro se ha demostrado que los fotorreceptores expresan NF-kappa-B de forma constitutiva. La exposición a la luz de los fotorreceptores produce un descenso en los niveles de NF-kappa-B tanto de la fracción nuclear como citosólica. Este descenso parece estar mediado por caspasa 1 y conduce a la apoptosis de las células ya que la presencia de la subunidad Re1A del factor NF-kappa-B en el núcleo es fundamental para la protección de los fotorreceptores contra la apoptosis mediada por la vía oxidativa (30).
Sin embargo, la activación de caspasas no parece estar implicada en el proceso que conduce a la apoptosis de los fotorreceptores en ratones sometidos a niveles altos de luz ya que los inhibidores de caspasas no reducen dicha apoptosis. El proceso de apoptosis en este modelo parece estar mediado por un tipo de proteasas dependientes de calcio denominadas calpaínas (31).
Los datos encontrados en la bibliografía sobre los mecanismos que determinan la muerte por apoptosis de los fotorreceptores son a veces contradictorios, parece que la activación de un tipo u otro de caspasas o incluso la activación de distintas vías depende del modelo experimental utilizado. Incluso utilizando el mismo modelo de daño, los resultados son a veces distintos. Un ejemplo claro es el que encontramos en dos trabajos que analizan los mecanismos que desencadenan la muerte de fotorreceptores durante el desprendimiento de retina. Según unos autores la apoptosis de los fotorreceptores en rata estaría mediada por la activación de caspasas (32); mientras que otros autores demuestran que la inhibición de las caspasas no reduce la muerte por apoptosis de los fotorreceptores en dicho modelo y concluyen que el proceso de apoptosis estaría mediado por vías dependientes de mitocondria pero no de caspasas. En concreto, demuestran que el factor inductor de apoptosis (AIF), un factor apoptótico independiente de caspasas, se relocaliza desde la mitocondria al núcleo (33).
En cuanto a las sustancias que bloquean la muerte por apoptosis de los fotorreceptores se ha demostrado, utilizando neuronas de la retina de rata en cultivo, que la adición de un ácido graso, el ácido docosahexaenoico, reduce la apoptosis de dichas células, preservando la integridad de la membrana mitocondrial y aumentando la expresión de Bcl-2 (34).
Otro mecanismo de neuroprotección de los fotorreceptores frente al daño inducido por la luz requiere la activación del receptor de insulina que produce la activación de PI3K en los segmentos externos de los bastones aislados de retinas de ratas y ratones adaptados a condiciones de luz (35). Este efecto neuroprotector ha sido también demostrado en fotorreceptores bovinos in vitro (36).
Apoptosis de las células ganglionares
Las vías de señalización que conducen a la apoptosis de las células ganglionares en la retina han sido analizadas utilizando distintos modelos experimentales de daño como la axotomía, la isquemia, la administración de sustancias excitotóxicas o el aumento de la presión intraocular.
Axotomía
La muerte de las células ganglionares de la retina tras la axotomía del nervio óptico en la rata se produce mediante apoptosis. Cuando se aplica un vector adenoviral que contiene el inhibidor de caspasas de mamíferos, XIAP (inhibidor de la apoptosis ligado al cromosoma X), en el nervio óptico de ratas sometidas a axotomía, de forma que sólo las células ganglionares lesionadas pueden transducirlo, se observa que XIAP inhibe la apoptosis de las células axotomizadas sugiriendo que dichas células degeneran a través de la actividad de las caspasas (37). Concretamente, la caspasa 3 y la caspasa 9, parecen estar implicadas en la muerte por apoptosis de las células ganglionares de la retina tras la axotomía del nervio óptico en la rata (38,39). Así, se ha demostrado que la administración de un inhibidor de la caspasa 3 o de un inhibidor de la caspasa 9 reducen la muerte de las células ganglionares (38-40) y producen una disminución en el número de microglía reactiva en la retina tras axotomía (41).
Además de la activación de las caspasas, se ha descrito que la axotomía del nervio óptico en la rata produce una disminución en los niveles de RNAm que codifican para dos moléculas antiapoptóticas, Bcl-2 y Bcl-XL (41).
Recientemente, se ha demostrado que el citocromo c juega un papel importante en la apoptosis de las células ganglionares de la retina tras axotomía. En la retina normal los niveles de citocromo c son bajos mientras que un día después de la axotomía el citocromo c aumenta, alcanzando un máximo a los tres días y disminuyendo después. El citocromo c se localiza principalmente en las células ganglionares por lo que estaría relacionado con la muerte de estas células (42).
Otra molécula que se activa y conduce a la apoptosis de las células ganglionares tras la axotomía es c-Jun. Los ratones con una mutación en el sitio de fosforilación de c-Jun presentan una menor muerte de células ganglionares de la retina tras la axotomía que los ratones sin dicha mutación (43). Por otro lado, se ha demostrado que tras la axotomía del nervio óptico, Hrk aumenta en la retina. Hrk es un regulador de la muerte celular que induce apoptosis en tejidos nerviosos. La expresión de este regulador se observa en una subpoblación de células ganglionares de la retina axotomizadas (44). Además, una proteína perteneciente a la familia de las MAPK, p38, se activa en el núcleo de las células ganglionares de la retina tras axotomía del nervio óptico. La administración del inhibidor de p38, SB203580, reduce la muerte de las células ganglionares de la retina (45).
Aunque la utilización de inhibidores de las caspasas parece inhibir la apoptosis de las células ganglionares tras la axotomía, estas sustancias no logran rescatar a las células de forma completa o duradera. En este sentido se ha demostrado que, aunque el tratamiento con el inhibidor de las caspasas se realice durante tiempos largos, no se aumenta su efecto protector (46).
Algunos factores tróficos pueden reducir la muerte por apoptosis de las células ganglionares tras la axotomía. En un modelo experimental en la rata, se ha demostrado que la administración del factor de crecimiento de la insulina (IGF-I) reduce la actividad de la caspasa 3 y previene la muerte de las células ganglionares de la retina. La vía de señalización, iniciada por IGF, que conduce a la supervivencia de las células ganglionares está mediada por la fosforilación de PI3K y Akt (47).
Otra neurotrofina que reduce la apoptosis de las células ganglionares de la retina tras la axotomía es el BDNF. El efecto protector del BDNF se produce mediante la reducción de la actividad de caspasa 9 (39) y de caspasa 3 (48). Además, el BDNF protege a las células ganglionares de la retina a través de un mecanismo dependiente de PI3K (48).
El efecto protector del BDNF ha sido puesto de manifiesto también en gato, cuando este factor neurotrófico se administra junto al factor neurotrófico ciliar (CNTF) y forskolina, se observa una disminución en la apoptosis de las células ganglionares de tipo beta, que son las más susceptibles a la muerte tras la axotomía del nervio óptico en esta especie (49).
El RNAm que codifica para el receptor de BDNF, TrkB, se reduce tras la axotomía del nervio óptico en la rata por lo que la transferencia del gen para TrkB en células ganglionares de la retina junto con la administración de BDNF exógeno aumenta la supervivencia de las células ganglionares de la retina hasta un 76% dos semanas después de la axotomía (50).
A diferencia del efecto neuroprotector del IGF, el efecto del BDNF no se bloquea tras la inhibición de la vía PI3K/Akt, lo que sugiere que el efecto inhibidor de apoptosis del BDNF no depende sólo de una vía de señalización intracelular (48). Recientemente se ha puesto de manifiesto que la administración en el vítreo de BDNF produce la activación de dos vías de señalización que conducen a la supervivencia celular, la vía de la MAPK y la vía de PI3K/Akt. La fosforilación de MAPK se produce en células ganglionares y células de Müller, mientras que Akt sólo se activa en las células ganglionares tras la administración de BDNF. Por lo tanto, parece que la colaboración de las dos vías es necesaria para que el efecto neuroprotector del BDNF se produzca (51). Sin embargo otros autores observan una activación de la vía de las MAPK pero no de la PI3K tras la administración de BDNF (50).
Utilizando la axotomía como modelo de daño a las células ganglionares de la retina se ha demostrado que, además de los factores tróficos, otras sustancias pueden tener un efecto neuroprotector sobre las células ganglionares de la retina. Así la administración de antibióticos como cicloheximida o actinomicina D reduce la muerte de células ganglionares de la retina inducida mediante axotomía en ratas (40). Por otro lado se ha demostrado que la administración en el vítreo de monosialogangliosido (GM1), sustancia que parece modular la respuesta neuronal a los factores tróficos, antes de la axotomía del nervio óptico en la rata, inhibe la degeneración de las células ganglionares de la retina axotomizadas y aumenta la activación de MAPK y de CREB, un factor de transcripción que es proteína de unión al elemento de respuesta a AMPc (52).
La sobreexpresión de moléculas antiapoptóticas, como Bcl-2 también logra rescatar a las células ganglionares de la retina de la muerte inducida por axotomía (53). En este sentido, utilizando la proteína HIV TAT que puede unir macromoléculas y transportarlas a través de membranas celulares, se consigue la entrada de Bcl-XL en las neuronas. Una vez dentro de las células, TAT-Bcl-XL es estable durante muchos días y mantiene su función, bloqueando la apoptosis de las células ganglionares de la retina inducida tras lesión en el nervio óptico en ratón (54).
La sobreexpresión de moléculas antiapoptóticas como Bcl-2 produce, no sólo una disminución en la apoptosis, sino un incremento en la regeneración del tracto óptico tras una lesión tectal en ratones neonatos transgénicos que sobreexpresan Bcl-2 (55). Este efecto, sin embargo, no se observa en los mismos animales tras pinzamiento del nervio óptico (56).
Utilizando un modelo de daño en el nervio óptico en ratón se ha analizado el papel de las calpaínas en el proceso de apoptosis que conduce a la muerte de las células ganglionares. El daño del nervio óptico produce, en un primer momento, una alteración en el transporte retrógrado y una acumulación y defosforilación de neurofilamentos en los axones. A más largo plazo se observa una pérdida de neurofilamentos en los axones degenerados. El análisis de la activación de calpaínas demuestra que estas proteasas puede contribuir al daño intraaxonal progresivo en las primeras etapas del daño y en la posterior degeneración axonal (57).
Por último, se ha demostrado que las semaforinas moléculas que en situación fisiológica guían el axon, son capaces de mediar la apoptosis en cultivos de neuronas. In vivo, se ha observado que tras la axotomía del nervio óptico en la rata se induce la expresión de proteínas semaforinas tipo III en la retina. Este aumento tiene lugar antes de que se produzca la apoptosis de las células ganglionares de la retina y parece estar implicado en este proceso ya que la inyección en el vítreo de un péptido derivado de la semaforina, sema3A, induce la perdida de células ganglionares de la retina y la pérdida de células ganglionares de la retina se inhibe en ojos axotomizados cuando se tratan con inyección de anticuerpos que bloquean la función de sema3A (58).
Daño excitotóxico
El aumento de glutamato en el vítreo que se produce como consecuencia de diferentes patologías oculares, puede producir un daño excitotóxico en la retina. Algunos trabajos han abordado el estudio de los mecanismos que conducen a la apoptosis de las células de la retina utilizando modelos de daño excitotóxico. Parece que el efecto apoptótico del glutamato en las células ganglionares estaría mediado por los receptores NMDA (N-metil-D-aspartato).
Se ha demostrado que la vía de las caspasas está implicada en la muerte por apoptosis de las células ganglionares de la retina tras exposición a dosis altas de glutamato. En concreto, la caspasa 3 se activa en las células de la capa de las células ganglionares y en las células de la capa nuclear interna de los explantes de retina de rata expuestos a dosis altas de glutamato. Esta activación precede a la aparición de las células con fragmentación de ADN en estas capas. Parece que la caspasa 1 no estaría implicada ya que un inhibidor de esta caspasa, el YVAD-CHO, no reduce la muerte de las células ganglionares de la retina en este modelo (59). Por otro lado, el calcio intracelular parece jugar un papel importante en la apoptosis de las células ganglionares de la retina inducida por glutamato ya que la nilvadipina, un bloqueante de los canales de calcio, bloquea el aumento en la concentración intracelular de este catión y reduce la apoptosis producida por glutamato (60). Además del calcio, c-Jun y la proteína reguladora p53 coexpresan en células ganglionares de la retina en cultivos organotípicos de retina de rata tratados con glutamato, lo que sugiere que ambas moléculas están implicadas en los mecanismos que conducen a la muerte de las células ganglionares de la retina en este modelo experimental de daño excitotóxico (61).
La activación de la vía de las caspasas también parece estar implicada en la muerte por apoptosis de las células de la retina en otro modelo de daño excitotóxico. Tras la inyección intravitreal de NMDA, un agonista de los receptores de glutamato, en el ojo de conejos adultos se ha observado que la apoptosis de las células en la capa de las células ganglionares y en la capa nuclear interna está mediada por caspasa 1 (62). p53 también está implicada en la regulación de la apoptosis mediada por NMDA. Tras la inyección intravitreal de NMDA en ratones normales y p53 deficientes se observa que, en los ratones normales, los niveles de RNAm que codifica para p53 están elevados. Este incremento se correlaciona con el comienzo de los cambios en la morfología de los núcleos de las células ganglionares de la retina. Los ratones deficientes en los dos alelos del gen p53 no muestran diferencias en el numero de células ganglionares de la retina después de la inyección de NMDA mientras que los ratones p53+/- tienen una pérdida menor. El análisis inmunohistoquímico muestra que un sustrato de las caspasas se localiza en los ratones p53+/+ y p53+/- pero no en los p53-/-. Parece que el NMDA provoca la muerte de células ganglionares de la retina mediante una vía dependiente de p53, sin embargo la presencia de p53 no es absolutamente necesaria puesto que los ratones que no poseen el gen presentan muerte celular a través de alguna vía alternativa. El análisis de la escisión del sustrato demuestra que la vía dependiente de p53 utiliza la vía de las caspasas mientras que la vía independiente de p53 no utiliza este mecanismo (63).
Recientemente se ha analizado el papel de la MAPK, p38, y de la vía de señalización PI3K/Akt en ratas tratadas con NMDA. Los animales fueron sometidos a inyección intravitreal de NMDA y de los inhibidores SB203580 (inhibidor de p38) y LY294002 (inhibidor de PI3K). El NMDA conduce a la muerte por apoptosis de las células ganglionares de la retina, antes de dicha apoptosis se observa un aumento en p38 y Akt en la capa de las células ganglionares y en la capa nuclear interna. SB203580 disminuye la muerte de las células ganglionares de la retina mientras que LY294002 la aumenta. Por lo tanto las vías de p38 MAPK y PI3K/Akt están implicadas en la señalización de la apoptosis inducida con NMDA. La vía de p38 es proapoptótica mientras que la vía de la PI3K es antiapoptótica (64).
Isquemia
La isquemia en la retina produce la muerte por apoptosis de las células a través de la activación de la vía de las caspasas. Sin embargo el tipo de caspasas que se activan en este proceso parece ser diferente en los distintos tipos celulares. Así, la expresión de caspasa 2 aumenta principalmente en la capa nuclear interna y en la capa de las células ganglionares mientras que la caspasa 3 aumenta principalmente en la capa nuclear interna y en la nuclear externa (65).
Además de la caspasa 2, la expresión de c-Jun aumenta en las células de la capa de las células ganglionares tras la isquemia (66). Otros autores han descrito la presencia de caspasa 3 en la capa nuclear interna y de caspasa 1 en la capa nuclear externa tras la isquemia (67). Utilizando el mismo modelo experimental Sumioka et al (68) demostraron que, además de la activación de caspasa 3 en las capas internas de la retina, la isquemia induce la aparición de un fragmento de 55 kDa que corresponde al fragmento activo de la proteína quinasa PKN. La aparición de esta proteína depende de la duración de la reperfusión y está relacionada con la muerte celular. En este sentido demuestran que la inhibición de la caspasa 3 reduce la aparición de PKN y disminuye la muerte de células en la retina.
Se ha demostrado que en ratas, un período corto de isquemia retiniana, anterior a otro período de isquemia más largo protege a la retina contra el daño. Parece que la liberación de adenosina, la síntesis de proteínas y mediadores como la proteína quinasa C (PKC) y canales de K+ dependientes de ATP, son necesarios para la protección. La isquemia precondicionante afecta a la expresión y activación de moléculas implicadas en los procesos de apoptosis, disminuyendo los cambios que se producen como consecuencia de la isquemia: activación de las caspasas 2 y 3, incremento de la expresión de Bax y PARP o activación de MAPK (69).
Glaucoma
El glaucoma es una patología extremadamente frecuente que produce la muerte por apoptosis de las células ganglionares de la retina de forma selectiva.
La vía de las caspasas está implicada en el proceso de apoptosis de las células ganglionares durante el glaucoma. En un modelo de experimental en la rata, se ha demostrado que el aumento crónico de la presión intraocular produce una activación de caspasa 3 en las células ganglionares de la retina. Además se observa un aumento en la escisión por parte de dicha caspasa de la proteína precursora amiloide (APP) (70).
La presencia de anticuerpos contra hsp27 ha sido descrita en enfermedades como el glaucoma. Además, se ha demostrado que la aplicación exógena de anticuerpos contra hsp27 promueve la muerte por apoptosis de las células. Tras su internalización, el anticuerpo hsp27 se detecta en estructuras citoplasmáticas y nucleares y colocaliza con el citoesqueleto de actina. La unión del anticuerpo hsp27 con la actina produce la depolimerización y escisión proteolítica de la actina y, por lo tanto, induce la apoptosis a través de la inhibición de la capacidad de hsp27 de estabilizar el citoesqueleto de actina. Esto sugiere que la presencia de autoanticuerpos contra hsp27 podrían reducir la supervivencia celular en patologías como el glaucoma (71).
EFECTO NEUROPROTECTOR DE FÁRMACOS HIPOTENSORES EN LA RETINA GLAUCOMATOSA
En los apartados anteriores hemos descrito algunas de las patologías oculares que desencadenan la apoptosis de las células de la retina. En la actualidad no existen tratamientos para enfermedades como la retinosis pigmentaria o el desprendimiento de retina que permita rescatar a las células de la muerte. Por el contrario, el glaucoma es una patología extremadamente frecuente que se trata con fármacos de distinta naturaleza, alguno de los cuales parece tener un efecto neuroprotector sobre las células ganglionares. En este apartado vamos a tratar de describir los trabajos encontrados en la bibliografía acerca del efecto neuroprotector de los fármacos utilizados en el tratamiento del glaucoma.
Uno de los principales factores de riesgo asociados con el glaucoma es el aumento crónico de la presión intraocular. En la actualidad, el glaucoma se diagnostica tras el seguimiento del paciente con presiones intraoculares altas, cuando el campo visual ya comienza a deteriorarse y, por lo tanto, cuando el proceso de la muerte de las primeras células ganglionares ha tenido lugar. Como único tratamiento del glaucoma se intenta disminuir la presión intraocular, bien farmacológicamente o bien de forma quirúrgica, cuando no hay una respuesta favorable al tratamiento farmacológico. Los agentes disponibles tienen efectos sobre la producción del humor acuoso, el drenaje convencional del humor acuoso a través de la trabécula y el drenaje no convencional a través de la raiz del iris y de la superficie del cuerpo ciliar.
Se ha demostrado que la brimonidina, un agonista de los receptores α2-adrenérgicos que reduce la presión intraocular a través de la disminución de la secreción de humor acuoso, reduce la muerte de las células ganglionares de la retina en un modelo experimental de isquemia (72-75), tras daño mecánico en el nervio óptico (76,77) o en un modelo de hipertensión ocular crónico en rata (77-79).
El efecto neuroprotector de la brimonidina se observa tanto cuando se administra por vía intraperitoneal como cuando se administra por vía tópica, y es dependiente de la dosis (76).
Se ha descrito que la brimonidina reduce los niveles de glutamato y aspartato en el humor vítreo tras isquemia en la rata (74). Además también reduce el incremento de inmunorreactividad para la proteína glial fibrilar (GFAP) que se produce en la retina tras el aumento en la presión intraocular (79).
Otros autores han sugerido que el efecto neuroprotector de la brimonidina estaría mediado por el aumento de BDNF en las células ganglionares; así, la inyección intravitreal de tartrato de brimonidina en ratas produce un aumento en el número de células ganglionares de la retina positivas a BDNF. Mediante Northern blot se demuestra un incremento del 28% en la expresión de BDNF en el grupo de brimonidina comparado con el grupo control (80). En un modelo de isquemia aguda en rata el efecto neuroprotector de la brimonidina sobre la retina parece estar mediado por un aumento en la expresión del factor de crecimiento de fibroblastos (bFGF), y de agentes anti-apoptóticos, como Bcl-2 y Bcl-XL, en la retina. Además este fármaco puede activar dos vías de señalización que determinan la supervivencia en la retina: la de las ERKs y la vía de la PI3K/Akt (81,82).
El nipradilol es otro fármaco utilizado en la clínica para el tratamiento del glaucoma (83), esta sustancia tiene un efecto antagonista no selectivo sobre los receptores ß-adrenérgicos y selectivo sobre los receptores α1-adrenérgicos. Además este compuesto tiene una acción vasodilatadora debido a la liberación de óxido nítrico mediada por la enzima intracelular glutation S transferasa (84). Se ha demostrado que el nipradilol tiene un efecto neuroprotector frente a la muerte de las células de la retina tras la administración de NMDA. Este efecto podría estar mediado por la liberación de óxido nítrico y la inhibición de la actividad de caspasa 3 (85). En este sentido, se ha demostrado utilizando cultivos de células PC12 que el efecto protector del nipradilol se reduce en presencia de un inhibidor de la proteína quinasa G (PKG), implicada en la vía del óxido nítrico (86).
Otro fármaco hipotensor es la unoprostona una sustancia sintética basada, al igual que el latanoprost, en la prostaglandina F2α. Este fármaco disminuye la presión intraocular mediante el aumento del flujo uveoescleral y tiene un efecto neuroprotector sobre las células ganglionares de la retina. La unoprostona inhibe la estimulación del glutamato y abre canales de potasio lo que produce una salida de este catión de las células y una hiperpolarización de las mismas. Como consecuencia de la hiperpolarización de las células se desencadena el cierre de canales de calcio dependientes de voltaje, disminuyendo el calcio intracelular y el daño neuronal (87).
Por último, se ha demostrado que un agonista adrenérgico selectivo de los receptores ß1, el betaxolol, reduce la entrada de sodio y calcio y protege a las neuronas de la retina durante el daño isquémico (88) y el daño inducido por glutamato durante el glaucoma (89). Otro mecanismo de protección del betaxolol parece ser el aumento en los niveles de mRNA para BDNF en la retina (88) y la reducción de la actividad de la isoenzima neuronal de la óxido nítrico sintasa (nNOS) en las células ganglionares de la retina observada tras isquemia-reperfusión (90).
BIBLIOGRAFÍA
1. Cohen JJ. Apoptosis. Immunol Today 1993; 14: 126-130. [ Links ]
2. Platt N, da Silva RP, Gordon S. Recognizing death: the phagocytosis of apoptotic cells. Trends Cell Biol 1998; 8: 365-372. [ Links ]
3. Galli-Resta L, Ensini M. An intrinsic time limit between genesis and death of individual neurons in the developing retinal ganglion cell layer. J Neurosci 1996; 16: 2318-2324. [ Links ]
4. Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science 1995; 267: 1456-1462. [ Links ]
5. Thornberry NA, Lazebnik Y. Caspases: enemies within. Science 1998; 281: 1312-1316. [ Links ]
6. Wolf BB, Green DR. Suicidal tendencies: apoptotic cell death by caspase family proteinases. J Biol Chem 1999; 274: 20049-20052. [ Links ]
7. Lincz LF. Deciphering the apoptotic pathway: all roads lead to death. Immunol Cell Biol 1998; 76: 1-19. [ Links ]
8. Jung Y, Miura M, Yuan J. Suppression of interleukin-1 beta-converting enzyme-mediated cell death by insulin-like growth factor. J Biol Chem 1996; 271: 5112-5117. [ Links ]
9. Troy CM, Stefanis L, Prochiantz A, Greene LA, Shelanski ML. The constrasting roles of ICE family proteases and interleukin-1beta in apoptosis induced by trophic factor withdrawal and by copper/zinc superoxide dismutase down-regulation. Proc Natl Acad Sci USA 1996; 93: 5635-5640. [ Links ]
10. Ahmad M, Srinivasula SM, Wang L, Talanian RV, Litwack G, Fernandes-Alnemri T et al. CRADD, a novel human apoptotic adaptor molecule for caspase-2, and FasL/tumor necrosis factor receptor-interacting protein RIP. Cancer Res 1997; 57: 615-619. [ Links ]
11. Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol 1998; 60: 619-642. [ Links ]
12. Hacker G. The morphology of apoptosis. Cell Tissue Res 2000; 301: 5-17. [ Links ]
13. Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res 1993; 57: 3976-3985. [ Links ]
14. Duband-Goulet D, Courvalin JC, Buendia B. LBR, a laminin and lamin binding protein from the inner nuclear membrane, is proteolyzed at late stages of apoptosis. J Cell Sci 1998; 111: 1441-1451. [ Links ]
15. Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol 2000; 10: 369-377. [ Links ]
16. Hengartner MO. The biochemistry of apoptosis. Nature 2000; 407: 770-776. [ Links ]
17. Jacobson MD, Raff MC. Programed cell death and Bcl-2 protection in very low oxygen. Nature 1995; 374: 814-816. [ Links ]
18. Hu Y, Benedict MA, Wu D, Inohara N, Nunez G. Bcl-XL interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc Natl Acad Sci USA 1998; 95: 4386-4391. [ Links ]
19. Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000; 403: 98-103. [ Links ]
20. Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science 1998; 281: 1305-1308. [ Links ]
21. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998; 94: 491-501. [ Links ]
22. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997; 91: 231-241. [ Links ]
23. Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signalling pathway by transcription-dependent and –independent mechanisms. Science 1999; 286: 1358-1362. [ Links ]
24. Tezel G, Wax MB. Inhibition of caspase activity in retinal cell apoptosis induced by various stimuli in vitro. Invest Ophthalmol Vis Sci 1999; 40: 2660-2667. [ Links ]
25. Chiarini LB, de Freitas FG, Leal-Ferreira ML, Tolkovsky A, Linden R. Cytoplasmic c-Jun N-terminal immunoreactivity: a hallmark of retinal apoptosis. Cell Mol Neurobiol 2002; 22: 711-726. [ Links ]
26. Wu J, Gorman A, Zhou X, Sandra C, Chen E. Involvement of caspase-3 in photoreceptor cell apoptosis induced by in vivo blue light exposure. Invest Ophthalmol Vis Sci 2002; 43: 3349-3354. [ Links ]
27. Yoshizawa K, Yang J, Senzaki H, Uemura Y, Kiyozuka Y, Shikata N, et al. Caspase-3 inhibitor rescues N -methyl N- nitrosourea-induced retinal degeneration in Sprague–Dawley rats. Exp Eye Res 2000; 71: 629-635. [ Links ]
28. Grimm C, Wenzel A, Hafezi F, Reme CE. Gene expression in the mouse retina: the effect of damaging light. Mol Vis 2000; 6: 252-260. [ Links ]
29. Yamazaki H, Ohguro H, Maeda T, Maruyama I, Takano Y, Metoki T, et al. Preservation of retinal morphology and functions in royal college surgeons rat by nilvadipine, a Ca(2+) antagonist. Invest Ophthalmol Vis Sci 2002; 43: 919-926. [ Links ]
30. Krishnamoorthy RR, Crawford MJ, Chaturvedi MM, Jain SK, Aggarwal BB, Al-Ubaidi MR et al. Photo-oxidative stress down-modulates the activity of nuclear factor-kappaB via involvement of caspase-1, leading to apoptosis of photoreceptor cells. J Biol Chem 1999; 274: 3734-3743. [ Links ]
31. Donovan M, Cotter TG. Caspase-independent photoreceptor apoptosis in vivo and differential expression of apoptotic protease activating factor-1 and caspase-3 during retinal development. Cell Death Differ 2002; 9: 1220-1231. [ Links ]
32. Zacks DN, Hanninen V, Pantcheva M, Ezra E, Grosskreutz C, Miller JW. Caspase activation in an experimental model of retinal detachment. Invest Ophthalmol Vis Sci 2003; 44: 1262-1267. [ Links ]
33. Hisatomi T, Sakamoto T, Goto Y, Yamanaka I, Oshima Y, Hata Y et al. Critical role of photoreceptor apoptosis in functional damage after retinal detachment. Curr Eye Res 2002; 24: 161-172. [ Links ]
34. Rotstein NP, Politi LE, German OL, Girotti R. Protective effect of docosahexaenoic acid on oxidative stress-induced apoptosis of retina photoreceptors. Invest Ophthalmol Vis Sci 2003; 44: 2252-2259. [ Links ]
35. Rajala RV, McClellan ME, Ash JD, Anderson RE. In vivo regulation of phosphoinositide 3-kinase in retina through light-induced tyrosine phosphorylation of the insulin receptor beta-subunit. J Biol Chem 2002; 277: 43319-43326. [ Links ]
36. Rajala RV, Anderson RE. Interaction of the insulin receptor beta-subunit with phosphatidylinositol 3-kinase in bovine ROS. Invest Ophthalmol Vis Sci 2001; 42: 3110-3117. [ Links ]
37. Kugler S, Straten G, Kreppel F, Isenmann S, Liston P, Bahr M. The X-linked inhibitor of apoptosis (XIAP) prevents cell death in axotomized CNS neurons in vivo. Cell Death Differ 2000; 7: 815-824. [ Links ]
38. Kermer P, Klocker N, Labes M, Thomsen S, Srinivasan A, Bahr M. Activation of caspase-3 in axotomized rat retinal ganglion cells in vivo. FEBS Lett 1999; 453: 361-364. [ Links ]
39. Kermer P, Ankerhold R, Klocker N, Krajewski S, Reed JC, Bahr M. Caspase-9: involvement in secondary death of axotomized rat retinal ganglion cells in vivo. Brain Res Mol Brain Res 2000; 85: 144-150. [ Links ]
40. Manabe S, Kashii S, Honda Y, Yamamoto R, Katsuki H, Akaike A. Quantification of axotomized ganglion cell death by explant culture of the rat retina. Neurosci Lett 2002; 334: 33-36. [ Links ]
41. Chaudhary P, Ahmed F, Quebada P, Sharma SC. Caspase inhibitors block the retinal ganglion cell death following optic nerve transection. Brain Res Mol Brain Res 1999; 67: 36-45. [ Links ]
42. Cheung ZH, Yip HK, Wu W, So KF. Axotomy induces cytochrome c release in retinal ganglion cells. Neuroreport 2003; 14: 279-282. [ Links ]
43. Yoshida K, Behrens A, Le-Niculescu H, Wagner EF, Harada T, Imaki J et al. Amino-terminal phosphorylation of c-Jun regulates apoptosis in the retinal ganglion cells by optic nerve transection. Invest Ophthalmol Vis Sci 2002; 43: 1631-1635. [ Links ]
44. Wakabayashi T, Kosaka J, Hommura S. Up-regulation of Hrk, a regulator of cell death, in retinal ganglion cells of axotomized rat retina. Neurosci Lett 2002; 318: 77-80. [ Links ]
45. Kikuchi M, Tenneti L, Lipton SA. Role of p38 mitogen-activated protein kinase in axotomy-induced apoptosis of rat retinal ganglion cells. J Neurosci 2000; 20: 5037-5044. [ Links ]
46. Kermer P, Klocker N, Bahr M. Long-term effect of inhibition of ced 3-like caspases on the survival of axotomized retinal ganglion cells in vivo. Exp Neurol 1999; 158: 202-205. [ Links ]
47. Kermer P, Klocker N, Labes M, Bahr M. Insulin-like growth factor-I protects axotomized rat retinal ganglion cells from secondary death via PI3-K-dependent Akt phosphorylation and inhibition of caspase-3 in vivo. J Neurosci 2000; 20: 2-8. [ Links ]
48. Klocker N, Kermer P, Weishaupt JH, Labes M, Ankerhold R, Bahr M. Brain-derived neurotrophic factor-mediated neuroprotection of adult rat retinal ganglion cells in vivo does not exclusively depend on phosphatidyl-inositol-3`-kinase/protein kinase B signalling. J Neurosci 2000; 20: 6962-6967. [ Links ]
49. Watanabe M, Fukuda Y. Survival and axonal regeneration of retinal ganglion cells in adult cats. Prog Retin Eye Res 2002; 21: 529-553. [ Links ]
50. Cheng L, Sapieha P, Kittlerova P. Hauswirth WW. Di Polo A. TrkB gene transfer protects retinal ganglion cells from axotomy-induced death in vivo. J Neurosci 2002; 22: 3977-3986. [ Links ]
51. Nakazawa T, Tamai M, Mori N. Brain-derived neurotrophic factor prevents axotomized retinal ganglion cell death through MAPK and PI3K signaling pathways. Invest Ophthalmol Vis Sci 2002; 43: 3319-3326. [ Links ]
52. Choi JS, Kim JA, Joo CK. Activation of MAPK and CREB by GM1 induces survival of RGCs in the retina with axotomized nerve. Invest Ophthalmol Vis Sci 2003; 44: 1747-1752. [ Links ]
53. Inoue T, Hosokawa M, Morigiwa K, Ohashi Y, Fukuda Y. Bcl-2 overexpression does not enhance in vivo axonal regeneration of retinal ganglion cells after peripheral nerve transplantation in adult mice. J Neurosci 2002; 22: 4468-4477. [ Links ]
54. Dietz GP, Kilic E, Bahr M. Inhibition of neuronal apoptosis in vitro and in vivo using TAT-mediated protein transduction. Mol Cell Neurosci 2002; 21: 29-37. [ Links ]
55. Chen DF, Schneider GE, Martinou JC, Tonegawa S. Bcl-2 promotes regeneration of severed axons in mammalian CNS. Nature 1997; 385: 434-439. [ Links ]
56. Lodovichi C, Di Cristo G, Cenni MC, Maffei L. Bcl-2 overexpression per se does not promote regeneration of neonatal crushed optic fibers. Eur J Neurosci 2001; 13: 833-838. [ Links ]
57. Saatman KE, Abai B, Grosvenor A, Vorwerk CK, Smith DH, Meaney DF. Traumatic axonal injury results in biphasic calpain activation and retrograde transport impairment in mice. J Cereb Blood Flow Metab 2003; 23: 34-42. [ Links ]
58. Shirvan A, Kimron M, Holdengreber V, Ziv I, Ben-Shaul Y, Melamed S et al. Anti semaphorin3A antibodies rescue retinal ganglion cells from cell death following optic nerve axotomy. J Biol Chem 2002; 277: 49799-49807. [ Links ]
59. Chen TA, Yang F, Cole GM, Chan SO. Inhibition of caspase-3-kike activity reduces glutamate induced cell death in adult rat retina. Brain Res 2001; 904: 177-188. [ Links ]
60. Otori Y, Kusaka S, Kawasaki A, Morimura H, Miki A, Tano Y. Protective effect of nilvadipine against glutamate neurotoxicity in purified retinal ganglion cells. Brain Res 2003; 961: 213-219. [ Links ]
61. Umihira J, Lindsey JD, Weinreb RN. Simultaneous expression of c-Jun and p53 in retinal ganglion cells of adult rat retinal slice cultures. Curr Eye Res 2002; 24: 147-159. [ Links ]
62. Kwong JM, Lam TT. N-methyl-D-aspartate (NMDA) induced apoptosis in adult rabbit retinas. Exp Eye Res 2000; 71: 437-444. [ Links ]
63. Li Y, Schlamp CL, Poulsen GL, Jackson MW, Griep AE, Nickells RW. p53 regulates apoptotic retinal ganglion cell death induced by N-methyl-D-aspartate. Mol Vis 2002; 8: 341-350. [ Links ]
64. Manabe S, Lipton SA. Divergent NMDA signals leading to proapoptotic and antiapoptotic pathways in the rat retina. Invest Ophthalmol Vis Sci 2003; 44: 385-392. [ Links ]
65. Singh M, Savitz SI, Hoque R, Gupta G, Roth S, Rosenbaum PS et al. Cell-specific caspase expression by different neuronal phenotypes in transient retinal ischemia. J Neurochem 2001; 77: 466-475. [ Links ]
66. Kurokawa T, Katai N, Shibuki H, Kuroiwa S, Kurimoto Y, Nakayama C et al. BDNF diminishes caspase-2 but not c-Jun immunoreactivity of neurons in retinal ganglion cell layer after transient ischemia. Invest Ophthalmol Vis Sci 1999; 40: 3006-3011. [ Links ]
67. Katai N, Yoshimura N. Apoptotic retinal neuronal death by ischemia-reperfusión is executed by two distinct caspase family proteases. Invest Ophthalmol Vis Sci 1999; 40: 2697-2705. [ Links ]
68. Sumioka K, Shirai Y, Sakai N, Hashimoto T, Tanaka C, Yamamoto M et al. Induction of a 55-kDa PKN cleavage product by ischemia/reperfusion model in the rat retina. Invest Ophthalmol Vis Sci 2000; 41: 29-35. [ Links ]
69. Zhang C, Rosenbaum DM, Shaikh AR, Li Q, Rosenbaum PS, Pelham DJ et al. Ischemic preconditioning attenuates apoptotic cell death in the rat retina. Invest Ophthalmol Vis Sci 2002; 43: 3059-3066. [ Links ]
70. McKinnon SJ, Lehman DM, Kerrigan-Baumrind LA, Merges CA, Pease ME, Kerrigan DF et al. Caspase activation and amyloid precursor protein cleavage in rat ocular hypertension. Invest Ophthalmol Vis Sci 2002; 43: 1077-1087. [ Links ]
71. Tezel G, Wax MB. The mechanisms of hsp27 antibody-mediated apoptosis in retinal neuronal cells. J Neurosci 2000; 20: 3552-3562. [ Links ]
72. Vidal-Sanz M, Lafuente MP, Mayor-Torroglosa S, Aguilera ME, Miralles de Imperial J, Villegas-Perez MP. Brimonidine´s neuroprotective effects against transient ischaemia-induced retinal ganglion cell death. Eur J Ophthalmol 2001; 11: 36-40. [ Links ]
73. Vidal-Sanz M, Lafuente MP, Mayor S, de Imperial JM, Villegas-Perez MP. Retinal ganglion cell death induced by retinal ischemia. Neuroprotective effects of two alpha-2 agonists. Surv Ophthalmol 2001; 45: S261-267. [ Links ]
74. Donello JE, Padillo EU, Webster ML, Wheeler LA, Gil DW. alpha (2)-Adrenoceptor agonists inhibit vitreal glutamate and aspartate accumulation and preserve retinal function after transient ischemia. J Pharmacol Exp Ther 2001; 296: 216-223. [ Links ]
75. Lafuente MP, Villegas-Perez MP, Mayor S, Aguilera ME, Miralles de Imperial J, Vidal-Sanz M. Neuroprotective effects of brimonidine against transient ischemia-induced retinal ganglion cell death: a dose response in vivo study. Exp Eye Res 2002; 74: 181-189. [ Links ]
76. Wheeler LA, Lai R, Woldemussie E. From the lab to the clinic: activation of an alpha-2 agonist pathway is neuroprotective in models of retinal and optic nerve injury. Eur J Ophthalmol 1999; 9: S17-21. [ Links ]
77. Wheeler LA, Woldemussie E. Alpha-2 adrenergic receptor agonists are neuroprotective in experimental models of glaucoma. Eur J Ophthalmol 2001; 11: S30-35. [ Links ]
78. Ahmed FA, Hegazy K, Chaudhary P, Sharma SC. Neuroprotective effect of alpha (2) agonist (brimonidine) on adult rat retinal ganglion cells after increased intraocular pressure. Brain Res 2001; 913: 133-139. [ Links ]
79. WoldeMussie E, Ruiz G, Wijono M, Wheeler LA. Neuroprotection of retinal ganglion cells by brimonidine in rats with laser-induced chronic ocular hipertension. Invest Ophthalmol Vis Sci 2001; 42: 2849-2855. [ Links ]
80. Gao H, Qiao X, Cantor LB, WuDunn D. Up-regulation of brain-derived neurotrophic factor expression by brimonidine in rat retinal ganglion cells. Arch Ophthalmol 2002; 120: 797-803. [ Links ]
81. Tatton WG, Chalmers-Redman RM, Sud A, Podos SM, Mittag TW. Maintaining mitochondrial membrane impermeability. An opportunity for new therapy in glaucoma? Surv Ophthalmol 2001; 45: S277-283. [ Links ]
82. Lai RK, Chun T, Hasson D, Lee S, Mehrbod F, Wheeler L. Alpha-2 adrenoceptor agonist protects retinal function after acute retinal ischemic injury in the rat. Vis Neurosci 2002; 19: 175-185. [ Links ]
83. Kanno M, Araie M, Koibuchi H, Masuda K. Effects of topical nipradilol, a beta blocking agent with alpha blocking and nitroglycerin-like activities, on intraocular pressure and aqueous dynamics in humans. Br J Ophthalmol 2000; 84: 293-299. [ Links ]
84. Adachi T, Hori S, Miyazaki K, Takahashi E, Nakagawa M, Udagawa A et al. Rapid increase in plasma nitrite concentration following intravenous administration of nipradilol. Eur J Pharmacol 1995; 286: 201-204. [ Links ]
85. Mizuno K, Koide T, Yoshimura M, Araie M. Neuroprotective effect and intraocular penetration of nipradilol, a beta-blocker with nitric oxide donative action. Invest Ophthalmol Vis Sci 2001; 42: 688-694. [ Links ]
86. Tomita H, Nakazawa T, Sugano E, Abe T, Tamai M. Nipradilol inhibits apoptosis by preventing the activation of caspase-3 via S-nitrosylation and the cGMP-dependent pathway. Eur J Pharmacol 2002; 452: 263-268. [ Links ]
87. Melamed S. Neuroprotective properties of a synthetic docosanoid, unoprostone isopropyl: clinical benefits in the treatment of glaucoma. Drug Exp Clin Res 2002; 28: 63-73. [ Links ]
88. Wood JP, DeSantis L, Chao HM, Osborne NN. Topically applied betaxolol attenuates ischaemia-induced effects to the rat retina and stimulates BDNF mRNA. Exp Eye Res 2001; 72: 79-86. [ Links ]
89. Zhang J, Wu SM, Gross RL. Effects of beta-adrenergic blockers on glutamate-induced calcium signals in adult mouse retinal ganglion cells. Brain Res 2003; 959: 111-119. [ Links ]
90. Cheon EW, Park CH, Kang SS, Cho GJ, Yoo JM, Song JK et al. Nitric oxide synthase expression in the transient ischemic rat retina: neuroprotection of betaxolol. Neurosci Lett 2002; 330: 265-269. [ Links ]


{kind=link}
{kind=link}
{kind=link}