










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versión impresa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.86 no.9 sep. 2011
Glaucoma hereditario asociado a displasia oculodentodigital
Hereditary glaucoma associated with oculodentodigital dysplasia
P. Tejadaa, Y.W. Eduardoa, E. Gutiérreza, A. Barcelóa y J. Sánchezb
aServicio de Oftalmología, Hospital Universitario 12 de Octubre, Madrid, España
bServicio de Pediatría, Unidad de Dismorfología, Hospital Universitario 12 de Octubre, Madrid, España
Dirección para correspondencia
RESUMEN
Caso clínico: Recién nacida de 20 días de vida con nistagmo ocasional, de madre con displasia oculodentodigital. En el examen físico se hallaban microcórneas veladas, nistagmo horizontal, tonometría de 35 en ojo derecho y 40mm Hg en el izquierdo, fondo de ojo normal; pirámide nasal y narinas estrechas y sindactilia de los dedos cuarto y quinto de ambas manos. Buena respuesta a trabeculectomía bilateral.
Discusión: La displasia oculodentodigital es una enfermedad hereditaria con marcada heterogeneidad fenotípica. La causa más frecuente de pérdida de visión es el glaucoma, siendo necesario su diagnóstico temprano con un seguimiento continuo y controles periódicos de la presión intraocular.
Palabras clave: Síndrome oculodentodigital. Glaucoma. Herencia.
ABSTRACT
Case report: A newborn evaluated at 20 days old due to occasional nystagmus. Her mother had presented with oculodentodigital dysplasia (ODDD) and glaucoma. The physical examination revealed opaque micro-corneas, and horizontal nystagmus. The tonometry showed 35mm Hg in OD and 40mm Hg in OS and the fundus examination was normal. She had a narrow nasal bridge with narrow nostrils, and fourth and fifth finger syndactylyl in both hands. A bilateral trabeculectomy was performed with a good response.
Discussion: ODDD is a rare autosomal dominant disease with heterogeneous phenotype manifestations. The most frequent cause of loss of visual acuity is the glaucoma, requiring long-term follow up with periodical control of the intraocular pressure (IOP).
Key words: Oculodentodigital síndrome. Glaucoma. Inheritance.
Introducción
La displasia oculodentodigital es una enfermedad hereditaria autosómica dominante que afecta al desarrollo normal del macizo facial, los ojos, los dientes y las extremidades, con una marcada heterogeneidad fenotípica intra e interfamiliar.
Las manifestaciones oftalmológicas más frecuentes son el microftalmos y la microcórnea, siendo más raros los casos con anomalías iridianas, cataratas y glaucoma secundario; las alteraciones dentarias incluyen la hipoplasia y la coloración amarillenta de los dientes, mientras que la sindactilia bilateral completa del cuarto y quinto dedo es la malformación más característica de las extremidades.
Caso clínico
Recién nacida a término que fue enviada para valoración oftalmológica a los 20 días de vida por la aparición de nistagmo ocasional. Su madre, sin antecedentes de importancia durante el embarazo, había sido operada de glaucoma a los 20 años de edad con trabeculectomía bilateral y cirugía reconstructiva de sindactilia de manos (Figs. 1 y 2). En el examen oftalmológico se observaban microcórneas con aspecto discretamente velado, nistagmo horizontal ocasional, tono ocular digital aumentado en forma bilateral y fondo de ojo (FO) con papilas de características conservadas; y el examen físico general revelaba una pirámide nasal estrecha con narinas estrechas y alas nasales finas, micrognatia leve y sindactilia de los dedos cuarto y quinto de ambas manos. Bajo sedación, la tonometría puso de manifiesto una presión intraocular (PIO) de 35 y 40mm Hg en los ojos derecho e izquierdo, respectivamente, y además se verificaron edema epitelial y nubéculas corneales superficiales.

Fig. 1. Microcórneas en la madre de la paciente.

Fig. 2. Resultado de la reconstrucción quirúrgica de
la sindactilia de manos en la madre de la paciente.
Se realizó una trabeculectomía bilateral con buen control post-operatorio de la PIO, sin necesidad de fármacos tópicos adicionales. En la última exploración oftalmológica, a los 14 meses de vida, se hallaba en ortotropia, con buena fijación y seguimiento de la luz, defecto refractivo de - 4 dioptrías en ambos ojos, ampollas difusas y sin signos inflamatorios (Figs. 3 y 4), y se confirmaba el buen control de la PIO. En el FO no se encontraron alteraciones de interés.

Fig. 3. Ojo derecho de la paciente con microcórnea.
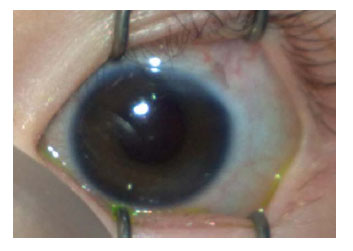
Fig. 4. Ojo izquierdo de la paciente con microcórnea,
ampolla plana y avascular.
Discusión
La displasia oculodentodigital es una enfermedad hereditaria muy rara que se transmite principalmente con un patrón autosómico dominante, siendo excepcionales los casos de herencia autosómica recesiva. Está causada por mutaciones heterocigóticas en el gen GJA1, localizado en el cromosoma 6q22-q23 y que codifica la proteína conexina 43 (Cx43), que parece desempeñar una función importante en el desarrollo ocular.
Se caracteriza típicamente por la alteración del desarrollo normal de la cara, los ojos, los dientes y las extremidades, además de alteraciones auditivas y neurológicas, aunque las manifestaciones clínicas son muy variables, incluso entre los miembros de una misma familia afectada.
La causa más frecuente de pérdida de visión es el glaucoma, aunque el tipo, la severidad y la edad de presentación son variables1; existiendo diversos mecanismos que lo condicionan, entre ellos alteraciones en el desarrollo del ángulo iridocorneal2, cambios gonioscópicos parecidos al glaucoma infantil y el glaucoma crónico de ángulo estrecho o abierto asociado a microcórnea3. Es importante destacar este último hecho, la asociación del glaucoma con microcórneas en edad infantil, como en nuestra paciente, ya que no siempre hallamos el clásico buftalmos. Además, se han descrito alteraciones genéticas que modifican el desarrollo normal del segmento anterior provocando una trabeculodisgenesia que se puede manifestar como un glaucoma congénito, como en nuestro caso, o en edad infantil1,4.
El diagnóstico temprano de este raro síndrome permitirá reconocer las alteraciones que puedan requerir terapia médica, e incluso quirúrgica, como el glaucoma congénito, que puede condicionar una mala visión e incluso la ceguera, y además permitirá el manejo multidisciplinario de las otras enfermedades asociadas con el fin de mejorar la calidad de vida del paciente.
En conclusión, resulta necesario conocer la posibilidad de desarrollo de glaucoma en este síndrome y la necesidad del seguimiento a largo plazo con controles periódicos de la PIO, ya que existe una gran variabilidad en la edad de presentación.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Bibliografía
1. Musa FU, Ratajczak P, Sahu J, Pentlicky S, Fryer A, Richard G, et al. Ocular manifestations in oculodentodigital dysplasia resulting from heterozygous missense mutation (L113P) in GJA1 (connexin 43). Eye advance online publication, 18 April 2008; doi:10.1038/eye.2008. [ Links ]
2. Judisch GF, Matin-Casal A, Hanson JW, Olin WH. Oculodentodigital dysplasia: four new reports and a literatura review. Ophthalmol. 1979; 97:878. [ Links ]
3. Traboulsi EI, Parks MM. Glaucoma in oculo-dento-osseous dysplasia. Am J Ophthalmol. 1990; 109:310-3. [ Links ]
4. Frasson M, Calixto N, Cronemberger S, de Aguiar RA, Leão LL, de Aguiar MJ. Oculodentodigital dysplasia: study of ophthalmological and clinical manifestations in three boys with probable autosomal recesive inheritance. Ophthalmic Genet. 2004; 25:227-36. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Correo electrónico: yurwil@gmail.com
(Y. W. Eduardo)
Recibido el 16 de Septiembre de 2010
Aceptado el 24 de Abril de 2011

