










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCCIÓN
El tumor maligno de la vaina nerviosa periférica (TMVNP), también llamado neurofibrosarcoma, schwannoma maligno o neurilemoma maligno, representa el 10 % de todos los sarcomas de tejidos blandos1.
Su localización más frecuente es en extremidades inferiores. Solo en torno al 10-20 % de las lesiones ocurren en la región de cabeza y cuello, siendo la mandíbula el sitio más comúnmente afectado1. Se encuentra asociado a neurofibromatosis en el 50 % de los casos, lo cual facilita su diagnóstico diferencial2.
Hasta donde sabemos, solo seis casos de tumor maligno de la vaina del nervio periférico (TMVNP) de la mandíbula sin asociación de neurofibromatosis han sido reportados en la literatura hasta la fecha.
CASO CLÍNICO
Acude a nuestra consulta un varón de 51 años derivado por su odontólogo para valorar una lesión radiolúcida mandibular identificada en la ortopantomografía (OPG) (Figura 1). Refería antecedentes médicos de diabetes mellitus tipo 2, dislipemia y depresión, sin hábitos tóxicos.
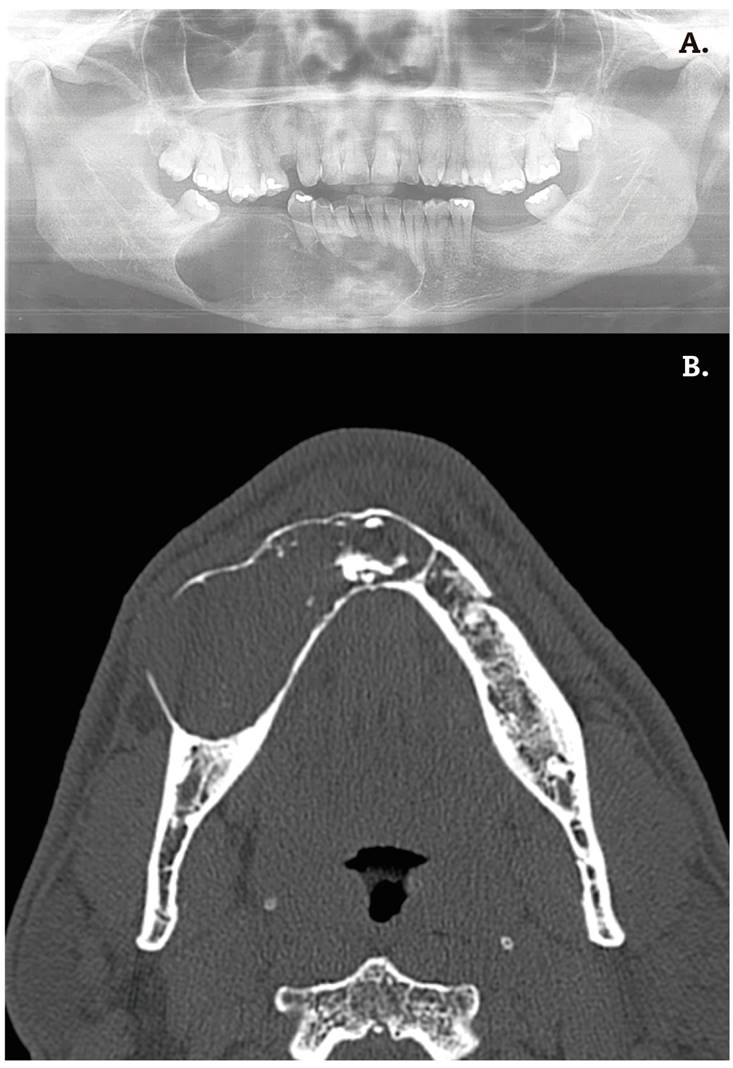
Figura 1. A. OPG: Lesión radiolúcida en cuarto cuadrante mandibular que abarca desde la cara mesial de las raíces de la pieza dentaria 47 hasta la cara distal apical de la pieza dentaria 33. No se identifica rizólisis de las raíces. B: TAC preoperatorio: lesión osteolítica con expansión de corticales en 4.º cuadrante.
A la exploración física se objetivó una tumoración en cuerpo mandibular derecho, con abombamiento de la cortical vestibular y lingual, sin lesiones intraorales. Presentaba movilidad de las piezas dentarias 43 y 44. No refería hipoestesia del nervio dentario inferior ni se palpaban adenopatías cervicales.
Se realizó una biopsia incisional con resultado compatible con pared de quiste dentario, negativo para células tumorales malignas. En la tomografía computarizada (TC) mandibular se evidenció una lesión osteolítica expansiva que se extendía hasta ocupar la práctica totalidad del cuarto cuadrante sin provocar rizólisis. La lesión presentaba áreas de calcificación grosera e incluía al nervio dentario inferior en su interior.
Ante los hallazgos descritos, el paciente fue intervenido para enucleación de la lesión y su posterior estudio histológico. Intraoperatoriamente, se visualizó una lesión con contenido sólido tumoral no quístico. La biopsia intraoperatoria fue informada como neoplasia mesenquimal de bajo grado. El resultado histológico definitivo informó de un TMVNP de bajo grado (Figura 2.A). El análisis inmunohistoquímico reveló positividad para S100, vimentina, CD34, Bcl2, PFGA, CD57, colágeno IV e histona (HH3) (Figura 2.B). Las células no fueron reactivas para panqueratina (AE1-AE3), actina HHF35, actina 1 A4, desmina, CD99, c-kit y melan-A. El MIB-1 fue entre el 1 y el 2 %.

Figura 2. Estudio microscópico. A. Anatomía patológica: proliferación celular que forma fascículos bien organizados e hipercelulares compuesto por células de morfología fusiforme con escasa atipia y marcado hipercromatismo nuclear. (HE, x200). B. Inmunohistoquímica: histona (HH3), x200.
El paciente fue reintervenido para resección con márgenes del tumor maligno. Se realizó una mandibulectomía segmentaria seguida de reconstrucción microquirúrgica con colgajo libre microvascularizado de peroné para una rehabilitación implantológica posterior. Se desarrolló una planificación con Materialise®, que incluía guías de corte mandibulares y de peroné, además de una placa de titanio impresa en 3D (Figura 3).
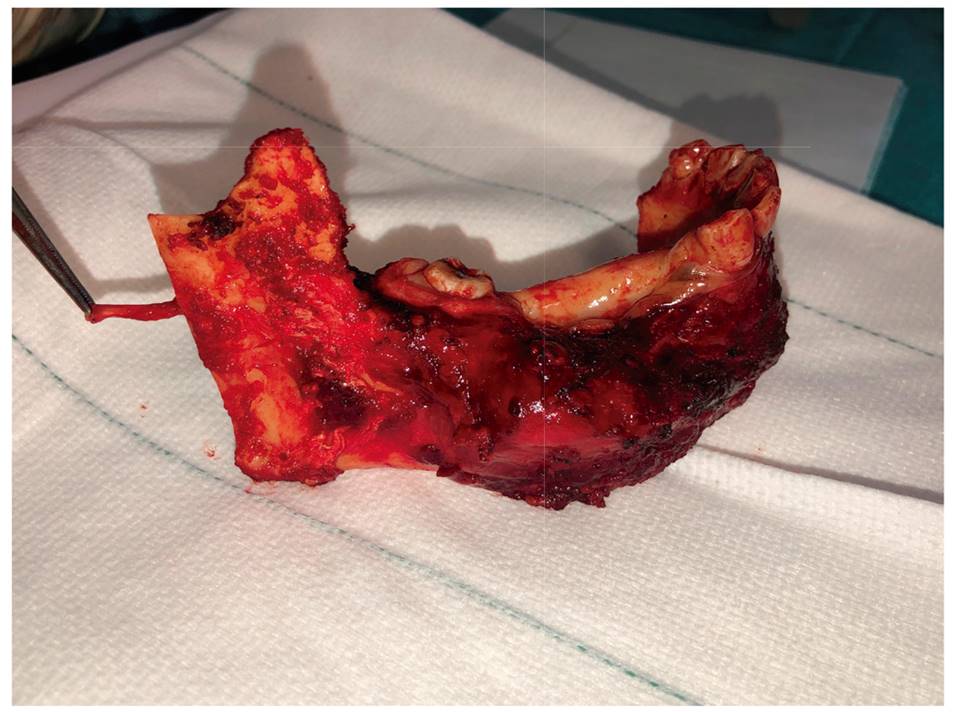
El estudio histológico de la pieza de resección confirmó el informe previo: neoformación maligna con fenotipo de tumor de vainas de nervio periférico, que infiltraba el hueso de forma masiva, sin afectación de ningún extremo de resección óseo o mucoso.
El caso fue discutido en la Unidad Multidisciplinar de Tratamiento de Tumores de Cabeza y Cuello del Hospital, decidiéndose administrar radioterapia adyuvante con dosis de 56 Gy.
Actualmente, tras seis meses de seguimiento, el paciente permanece asintomático y sin signos de recidiva tumoral (Figura 4).

DISCUSIÓN
Se realizó una búsqueda bibliográfica en PubMed con los términos libres ''malignant peripheral nerve sheath tumor'' AND ''jaw'' entre los años 2000-2019. Únicamente se identificaron ocho artículos en los que se describían casos de tumor maligno de vainas de nervio periférico, que afectaban el nervio dentario inferior.
El TMVNP es una neoplasia muy infrecuente, con una incidencia estimada de 0,001 % en la población general 2,3,4. Representa el 10 % de todos los sarcomas de tejidos blandos, de los cuales únicamente entre el 8 y el 16 % se localizan en la región anatómica de cabeza y cuello. Se trata de una entidad muy agresiva, cuyo origen deriva de las células de la vaina nerviosa: célula de Schwann, fibroblastos perineurales o fibroblastos endoneuriales. Genéticamente está relacionado con la pérdida de secuencia del brazo cromosómico 17q que provoca la inactivación del gen neurofibromatosis-11.
Casi en un 50 % de los casos, el TMVNP se encuentra asociado a neuroflbromatosis tipo I (NF1). Los pacientes con NF1 tienen hasta 4000 veces más riesgo de desarrollar TMVNP en el transcurso de sus vidas que los individuos que no presentan esta condición. La tasa de transformación maligna de un neurofibroma se calcula en 5-16 %2. Aun así, también se observan TMVNP desarrollados de novo. De igual modo, la radiación ha sido estudiada como un factor etiológico en el desarrollo de TMVNP, con un periodo de latencia de 10 a 20 años5. La gran mayoría de los TMVNP aparecen en el grupo de edad comprendido entre los 20 y los 50 años6, sin predilección por ninguna raza o sexo. En los casos asociados a NF1, la edad de aparición es incluso menor.
En los casos publicados, el TMVNP se manifiesta como una masa indolora de crecimiento progresivo, asociado a hipoestesia de la región inervada por el nervio dentario inferior. Radiológicamente se expresa con un patrón destructivo completo con expansión ósea, erosión y ensanchamiento del canal del nervio5,6,7,8. El diagnóstico se basa fundamentalmente en la histopatología de la lesión, y su confirmación mediante inmunohistoquímica, que refleja la diferenciación de la célula de Schwann. El TMVNP en ocasiones se asemeja a tejidos blandos no neurales, como neoplasias malignas de células fusiformes, incluyendo variantes de sarcoma sinovial, fibrosarcoma, leiomiosarcoma, melanoma metastásico y lesiones celulares reactivas óseas como tumor fibroso solitario y fascitis nodular. El TMVNP, sin embargo, tiene contornos irregulares con heterogeneidad morfológica, con núcleos ondulados y estroma mixoide, lo que sugiere un origen neurógeno1. El estudio inmunohistoquímico muestra una inmunorreactividad a la proteína S-100 y vimentina, con positividad focal a CD68 y negatividad a la queratina. Aunque S-100 no es exclusivo del TMVNP, sí es indicativo de diferenciación neural1,2,3.
Todos los autores coinciden en que el tratamiento de TMVNP es la resección quirúrgica amplia1,2,3,4,5,6,7,8,9. Para ello, la planificación 3D nos permite conseguir márgenes libres con mayor seguridad. El uso de la radioterapia es controvertido, pero estudios recientes han evidenciado la radiosensibilidad de este tumor, ofreciendo mejores tasas de control tumoral y supervivencia1. El uso de radioterapia como única modalidad de tratamiento es cuestionable, aunque puede estar indicada en pacientes no operables2. El rol de la quimioterapia en el tratamiento del TMVNP aún no está definido, aunque podría ser apropiada como terapia adyuvante en ciertos pacientes con enfermedad sistémica o metastásica.
El pronóstico del TMVNP es generalmente desfavorable. Presenta una alta tasa de recurrencia, entre 30-60 %, incluso tras resecciones locales completas. Su diseminación suele ser por extensión directa, vía hematógena o por diseminación perineural, siendo rara la diseminación linfática. La diseminación metastásica más frecuente es a pulmón y huesos1,3. La tasa de supervivencia global y libre de enfermedad a los cinco años es de 52 % y 33 %, respectivamente, teniendo peor pronóstico los pacientes con NF13,4. Aunque en la literatura no se identifica el seguimiento mínimo, en las curvas de supervivencia se produce una estabilización a los 5 años4. Los factores pronósticos son el tamaño de la lesión, la ubicación, el estadio y el grado histológico. A pesar de ello, actualmente dado el número tan limitado de casos y la falta de seguimiento a largo plazo, es difícil plantear un pronóstico fiable3.
En conclusión, el TMVNP es un tumor muy infrecuente pero altamente agresivo. Precisa un tratamiento con márgenes de seguridad y un seguimiento estrecho por su alta recurrencia.

