










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
versión impresa ISSN 1137-6627
Anales Sis San Navarra vol.35 no.2 Pamplona may./ago. 2012
https://dx.doi.org/10.4321/S1137-66272012000200010
Obesidad monogénica humana: papel del sistema leptina-melanocortina en la regulación de la ingesta de alimentos y el peso corporal en humanos
Monogenic human obesity: role of the leptin-melanocortin system in the regulation of food intake and body weight in humans
E. González Jiménez1, M.J. Aguilar Cordero2, C.A. Padilla López3, I. García García4
1. Departamento de Enfermería. Facultad de Enfermería (Campus de Melilla). Universidad de Granada. Melilla.
2. Departamento de Enfermería. Facultad de Ciencias de la Salud. Universidad de Granada. Melilla.
3. Grupo PAI de investigación CTS-367. Granada.
4. Departamento de Enfermería. Facultad de Ciencias de la Salud. Universidad de Granada. Melilla.
Dirección para correspondencia
RESUMEN
La obesidad humana es un trastorno de origen multifactorial en el que intervienen factores tanto genéticos como ambientales. La existencia de alteraciones genéticas que dan origen a obesidades monogénicas resulta muy interesante para el estudio de los mecanismos que contribuyen a un aumento de la ingesta de energía y la acumulación de grasa en el cuerpo. La mayoría de los genes implicados en obesidad monogénica se relacionan con el sistema de la leptina-melanocortinas, de ahí la importancia de su estudio a través de mutaciones naturales en ratones. Así, se han descrito mutaciones relacionadas con obesidad humana de tipo monogénica en la leptina y su receptor, proopiomelanocortina y prohormona convertasa 1. El objetivo de este trabajo ha sido ofrecer una revisión actualizada acerca de las principales características y funcionamiento del sistema leptina-melanocortinas, así como de sus implicaciones y potencialidades en el proceso de regulación de la ingesta alimentaria y control del peso corporal.
Palabras clave: Obesidad. Leptina. Melanocortinas. Proopiomelanocortina. MC4R.
ABSTRACT
Human obesity is a disorder of multifactorial origin in which genetic and environmental factors are involved. To understand the mechanisms regulating energy intake and fat accumulation in the body, it is important to study the genetic alterations causing monogenic obesity. Most of the genes involved in monogenic obesity are associated with the leptin-melanocortin system; hence the importance of studying this system by analysing natural mutations in mice. Previous studies have described mutations in leptin and its receptor, proopiomelanocortin and prohormone convertase 1 associated with human obesity of monogenic origin. The aim of this study is to provide an updated review of the main characteristics and functioning of the leptin-melanocortin system, and its implications and potentialities in regulating food intake and body weight.
Key words: Obesity. Leptin. Melanocortins. Proopiomelanocortin. MC4R.
Introducción
En la actualidad, la obesidad se ha convertido en un problema de salud pública a nivel mundial por su estrecha vinculación con las principales causas de morbilidad y mortalidad en países industrializados y en vías de desarrollo1. La obesidad se caracteriza por una acumulación de grasa a nivel corporal cuya fuente de origen estriba en un desequilibrio entre la ingesta calórica y el gasto energético del individuo2. A pesar de la indudable influencia de los factores socioculturales y ambientales en el desarrollo de la obesidad humana, la evidencia científica actual pone de manifiesto la importancia del componente genético en la génesis de este trastorno3. Teniendo en cuenta que la obesidad monogénica instituye la causa-efecto para el desarrollo de la obesidad, el análisis de estos defectos genéticos resultará relevante para el estudio y comprensión de los factores implicados en el proceso de homeostasis energética4.
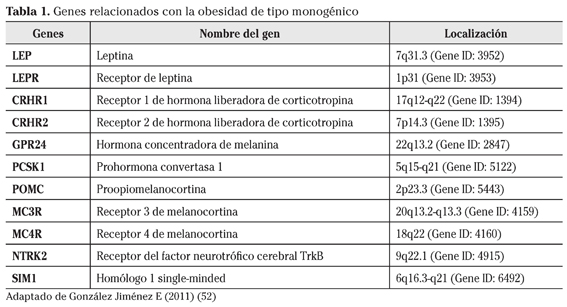
La obesidad de tipo monogénico tiene su origen en un único gen disfuncional y representa un pequeño número de casos que aparecen en la niñez y que generalmente suelen venir acompañados de diferentes trastornos neuroendocrinos, del desarrollo y la conducta5. En la actualidad se conocen numerosas de estas mutaciones las cuales asientan a menudo sobre genes relacionados con el control de la ingesta y con el mecanismo de acción de la leptina en el sistema nervioso central a través de la vía de las melanocortinas (Tabla 1).
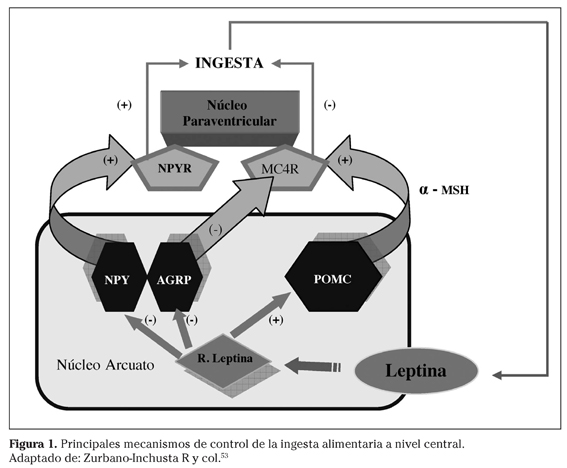
La leptina, hormona sintetizada y secretada fundamentalmente por el tejido adiposo blanco, circula en plasma en una concentración proporcional al volumen de grasa corporal6. Dicha hormona atraviesa la barrera hematoencefálica para actuar como una señal indicadora de las reservas energéticas interactuando con su receptor específico a nivel del núcleo arcuato del hipotálamo7. Dicho núcleo posee dos tipos de poblaciones neuronales con altos niveles de expresión del receptor de leptina: las neuronas POMC/CART, las cuales transfieren señales anorexigénicas a través de los derivados de la proopiomelanocortina (POMC), y las neuronas AGRP/NPY, que transfieren señales estimuladoras de la ingesta a través del neuropéptido Y (NPY) y la proteína relacionada con Agouti (AGRP)8. Ante situaciones de niveles reducidos de leptina (durante el ayuno prolongado o por deficiencia genética de leptina), se favorece la expresión de AGRP/NPY, lo que impulsa a una mayor ingesta de alimentos9. En aquellas situaciones de exceso de leptina, se promueve la expresión de POMC, que se separa en péptidos denominados melanocortinas (α-MSH y β-MSH) quienes a su vez actuarían como ligandos endógenos del receptor 4 de melanocortina (MC4R). Por el contrario, AGRP actuaría como un agonista inverso en este receptor, promoviendo la ingesta de alimentos10.
Alteraciones en los genes relacionados con el desarrollo hipotalámico
En relación con el desarrollo de obesidad en humanos, se ha logrado identificar anomalías en hasta tres genes asociados con el desarrollo del hipotálamo. Dichos genes son Sim1, BDNF y NTRK 11. Estos tres genes desarrollan funciones importantes durante el desarrollo y maduración del hipotálamo, si bien las causas exactas por las que sus mutaciones de asocian con el desarrollo de obesidad aún se desconocen11.
Sim 1
En el año 2000 fue descrito el primer caso en una niña con obesidad extrema de comienzo temprano, aceleración en su crecimiento y gasto energético normal por mutación en el gen Sim1 (6q16.3-q21)12. Dicha paciente no presentaba anomalías en su desarrollo, tampoco rasgos dimórficos, ni alteraciones endocrinológicas, mostrando únicamente una translocación de novo en uno de los alelos del gen Sim113.
Se ha estudiado en ratones con una única copia del gen Sim1 y presentan el mismo fenotipo que la paciente descrita y muestran una disminución en el número de neuronas de su núcleo paraventricular (imprescindibles para el balance energético y que expresan MC4R). Se plantea la hipótesis de que sea ésta la causa de obesidad en los ratones heterocigotos para Sim1 y en los pacientes con haploinsuficiencia para Sim114 en un estudio reciente demostró como el gen Sim1 está implicado en la regulación de la alimentación. Por otra parte, puso de manifiesto que la hiperfagia y con ello la obesidad en ratones deficientes de Sim1 podía ser debida a cambios en el sistema leptina-melanocortina-oxitocina15.
Factor neurotrófico derivado del cerebro (BDNF)
El factor neurotrófico derivado del cerebro (BDNF) y su receptor TRKB (tropomiosina relacionada con la quinasa B), participan en la regulación, proliferación, supervivencia y diferenciación de las neuronas durante su etapa de desarrollo, así como la plasticidad neuronal, memoria y desarrollo cognitivo en el sistema nervioso del adulto16. Asimismo, el BDNF interviene en el metabolismo energético y en la conducta alimentaria. En este sentido, deficiencias parciales de BDNF en modelos de ratón serán causa de hiperfagia y obesidad. El primer caso descrito por disrupción del gen BDNF (11p13) en humanos fue el de una niña de 8 años obesa, la cual presentaba una anomalía en el cromosoma 11 que alteraba el gen BDNF en uno de los puntos de rotura cromosómicos17.
La expresión de BDNF esta regulada por la señalización de MC4R en el hipotálamo ventromedial, donde se une a su receptor. Según esto, y atendiendo a resultados de estudios recientes cabe resaltar como la infusión cerebral de BDNF corrige la hiperfagia en los ratones deficientes de MC4R18.
Teniendo en cuenta todo lo anterior, un mayor y mejor conocimiento del sistema leptina-melanocortinas resultará esencial para la comprensión de los factores y procesos implicados en la regulación de la ingesta alimentaria en pacientes con obesidad.
Leptina y su receptor
En 1994, el genetista M. Jeffrey Friedman identificó el gen que codifica para la hormona leptina así como la mutación responsable de la obesidad e hiperfagia característica de los ratones ob19. La mutación, en estado homocigoto, se caracteriza por una sustitución C>T en la posición 105, que convierte un codón de arginina en un codón de terminación prematura19. Posteriormente, se demostró cómo la administración exógena de leptina generaba una disminución de la ingesta del ratón ob, disminuyendo su obesidad20. Sin embargo, cuando los defectos genéticos tienen lugar a nivel del receptor de la leptina (ratón db/db), (sustitución G>T en la posición 106 en estado homocigoto), el estado de obesidad no revierte con la administración exógena de leptina20.
Asimismo, se han descrito mutaciones del gen de la leptina en 5 familias de origen pakistaní y en una familia de origen turco 21. En todas ellas, la administración exógena de leptina humana disminuyó considerablemente el peso corporal y la ingesta de estos sujetos obesos21.
El receptor de la leptina (LEPR) es una proteína de membrana homóloga al receptor de las citoquinas tipo 1, del que existen diferentes isoformas generadas mediante ensamblaje alternativo22. La unión de la leptina a su receptor en el hipotálamo facilita la dimerización del receptor, lo que provoca una activación de las quinasas JAK2 las cuales posibilitan su autofosforilación y la fosforilación de residuos de tirosina en LEPR. Este proceso permite la activación de factores de transcripción (STAT3), que serán dimerizados y translocados hasta el núcleo23. Otros factores intervinientes serán SOCS3 y PTP1B quienes a través de la acción del JAC2 culminarán con el proceso de señalización. Todo ello en su conjunto hará posible la síntesis de péptidos anorexígenos23.
Por otra parte, se ha descrito que mutaciones en el gen que codifica el receptor de la leptina (P316:W646C), pueden ser fuente de hiperfagia y obesidad temprana. Así, Andiran y col (2011)24 describen el caso de una niña de 6 años de edad con obesidad monogénica debida a una deficiencia congénita t otal del receptor de la leptina.
Otra ruta alternativa de señalización activada por la leptina es la fosfatidil inositol 3 quinasa (PI3K). En ella se induce a la fosforilación de los residuos de la tirosina en el sustrato del receptor insulínico (IRS2) por mediación de JAK2, quienes a su vez fosforilan y activan PI3K25. Por otro lado, la proteína SH2B se adhiere a JAK2 e IRS2, permitiendo la activación de la ruta PI3K. Sujetos con una deficiencia a nivel de este gen desarrollarían resistencia a la insulina, a la leptina, así como hiperfagia y obesidad26. Por último, la ruta PI3K será activada por unión de la insulina a su receptor, circunstancia que completa la confluencia de señalización entre estas dos hormonas intervinientes en la homeostasis energética27.
Mutaciones que afectan al sistema leptina-melanocortinas
Mutaciones que afectan a los receptores 3 y 4 de melanocortinas
El sistema de las melanocortinas está compuesto por la proopiomelanocortina (POMC) y los péptidos derivados de POMC que actúan como ligandos de los receptores de melanocortinas: α-, β- y γ-MSH (Hormonas α, β- y y estimulantes de melanocitos), así como la hormona adrenocorticotropa (ACTH). En segundo lugar, por una familia de cinco receptores de melanocortina (MC1R-MC5R). Por último, los péptidos que antagonizan el efecto de los ligandos derivados de POMC: AGRP (proteína relacionada con agouti) y ASIP (proteína señalizadora de agouti)28.
Los receptores de melanocortinas se encuentran adheridos a proteínas G, siendo expresados en regiones del sistema nervioso central las cuales se relacionan con el control del apetito. Según esto, ratones con deficiencia genética de algunos de estos receptores desarrollarán obesidad e hiperfagia28. En humanos, las mutaciones en alguno de ellos, al igual que en el ratón, serán causa de obesidad de tipo monogénico28. Actualmente, la prevalencia mundial de obesidad asociada a mutaciones en MC4R se estima en un 2,5%29. En 1998, se publicaron los dos primeros casos de obesidad en humanos por mutaciones en el gen MC4R (18q22). Todos los pacientes evolucionaron clínicamente con hiperfagia y obesidad. Los efectos mediados por MC4R suelen desarrollarse a través de la estimulación de las vías anorexigénicas e inhibición de las rutas neuronales orexigénicas29.
Marti y col30, analizando la posible existencia de mutaciones en el gen MC4R en población española, encontraron una nueva mutación en el codón 16 (Trp16Stop) en una mujer obesa. Del mismo modo, aislaron la mutación (Val253Ile) previamente descrita en un sujeto no obeso, el polimorfismo Val103Ile en otro individuo obeso y el polimorfismo Ile251Leu con igual prevalencia entre personas obesas y no obesas.
La mayoría de las mutaciones MC4R son heterocigotas heredadas de forma dominante, aunque han sido descritos casos de homocigosidad o heterocigosidad compuesta con un patrón autosómico recesivo. Actualmente no existe tratamiento alguno para estas anomalías. Si bien, estudios recientes muestran cómo la recuperación de expresión en la superficie celular de mutantes MC4R podría tener un beneficio terapéutico dado que las mutaciones MC4R causantes de obesidad, inducen a una retención intracelular de receptores mediante un sistema de control de calidad celular31.
Por su parte, el receptor de melanocortina-3 (MC3R), será otro importante regulador de la homeostasis energética. Estudios en roedores han demostrado que dicho receptor, al igual que el receptor de melanocortina-4 (MC4R), es esencial en la regulación del balance energético32. Si bien, mientras que las mutaciones de MC4R se han establecido firmemente como una de las causas de la obesidad monogénica, existe cierta controversia sobre si las mutaciones en el gen MC3R guardan relación con la patogénesis de la obesidad humana32.
Del mismo modo, estudios recientes han identificado el gen SH2B1 por su potencial implicación en el desarrollo de obesidad. Así en el estudio desarrollado por Bochukova y col (2010)33 se pudo comprobar como el locus de dicho gen se asociaba significativamente con el desarrollo de obesidad dada su estrecha implicación en el proceso de señalización de la leptina e insulina.
Mutaciones que afectan al gen de proopiomelanocortina
Hasta el momento, en humanos se han identificado 3 mutaciones diferentes en el gen POMC, concretamente en dos pacientes pediátricos alemanes. Una niña heterocigoto, portadora de dos mutaciones en el exón 3 de las proteínas (G7013T y C7133X) las cuales interfieren con el procesamiento postraduccional de la poliproteína POMC y disminuyen la concentración de sus derivados: ACTH; α-MSH y β-endorfina34. La segunda mutación la presentó un paciente homocigótico masculino portador de una alteración a nivel de la C3804A del exón 2, alteración que cursa con la inhibición total de la transcripción de POMC. Ambos pacientes manifestaron un fenotipo de obesidad durante los primeros meses de vida, acompañados de pigmentación pelirroja en el cabello e insuficiencia adrenal34. En el caso del ratón deficiente en POMC, éste desarrolla hiperfagia, obesidad, pigmentación alterada, hiperleptinemia, y ausencia de hormona adrenocorticotropa34.
Mutaciones que afectan a los enzimas convertasas PCSK1 y PCSK2
Otras biomoléculas implicadas son los enzimas convertasas 1 y 2 (PCSK1 y PCSK2), las cuales actúan separando POMC en un proceso postraduccional que resulta diferente en el hipotálamo con respecto a la hipófisis35. Se ha descrito deficiencia genética de PCSK1 en dos casos de obesidad con mutaciones en estado de heterocigoto compuesto. Por el contrario, el ratón deficiente en esta enzima no presenta un claro fenotipo de obesidad, caracterizándose por un tamaño corporal reducido y defectos endocrinos como niveles indetectables de insulina y altos niveles de proinsulina36. Por lo tanto, la obesidad humana por deficiencia de PCSK1 parece tener mayores similitudes con el modelo de ratón obeso fat/fat generado por la ausencia de carboxipeptidasa E que con la deficiencia genética de PCSK1 en el ratón36.
Sistema leptina-melanocortinas y su conexión con otros mecanismos reguladores
Estudios experimentales han puesto de manifiesto que la supresión del receptor de leptina en neuronas POMC/CART genera una obesidad de menor magnitud que la descrita en el ratón db/db37. Ello es una muestra de la importancia de la leptina y su receptor37 más allá de su acción a través del núcleo arcuato (Fig. 1). Por otra parte, siguiendo con el sistema leptina-melanocortinas cabe destacar como los axones de las neuronas POMC/CART y AGRP/NPY se proyectan hacia otras regiones hipotalámicas igualmente importantes en el proceso de regulación de la ingesta, tales como el núcleo paraventricular, el área hipotalámica lateral, el núcleo ventromedial y el núcleo dorsomedial, que expresan MC4R. Asimismo, se ha descrito igualmente la expresión de MC4R en el núcleo del tracto solitario, donde actuaría sobre el efecto de saciedad inducido por factores generados en el intestino38.
Recientemente, se ha propuesto que antagonistas sintéticos de MC4R podrán bloquear el efecto de saciedad inducido por el neurotransmisor nesfatina, el cual inhibe el apetito y disminuye la cantidad de grasa en el organismo, sin causar daño a la masa muscular39.
La unión de ligandos de melanocortinas a MC4R provoca una inhibición de la ingesta mediante mecanismos que parecen incluir al factor neurotrófico cerebral BDNF, una proteína que, en los seres humanos, es codificada por el BDNF40. Entre las funciones de esta neurotrofina destacan producir diferenciación de las células madre o stem cell neuronales, inducir al crecimiento, diferenciación neural y sináptica y participar en fenómenos plásticos como la remodelación sináptica, el aumento de las conexiones sinápticas y la diferenciación y crecimiento neural40.
En ratones, la deleción del gen homólogo al BDNF humano en los núcleos hipotalámicos ocasiona hiperfagia y con ello obesidad41. En humanos, el importante papel de las neurotrofinas se ha manifestado en un caso de obesidad severa con hiperactividad y deficiencia cognitiva, caracterizado por una insuficiencia de BDNF, así como por la obesidad e hiperfagia por la pérdida de un segmento cromosómico que contiene al gen BDNF en algunos pacientes afectados con el síndrome WAGR, causado por una mutación en el cromosoma 11, concretamente en la región 11p1342.
También se han descrito casos de obesidad en modelos de ratón caracterizados por hiperfagia y retraso mental ante deficiencia parcial de BDNF y TRKB43. El primer caso descrito en humanos por disrupción del gen BDNF (11p13,OMIM#113505) se trataba de una niña de 8 años que presentaba un cuadro de obesidad e hiperfagia severa44, la cual portaba una inversión paracéntrica de novo en el cromosoma 11 que alteraba el gen BDNF. A pesar de que dicha inversión podría alterar otros genes, la gran similitud de esta paciente con el primero descrito con una mutación en el gen NTRK2 (9q22.1, OMIM#600456), el gen que codifica el receptor de BDNL, TRKB45, reafirma la hipótesis de que su fenotipo era causado por haploinsuficiencia de BDNF.
Sistema leptina-melanocortinas y su importancia en el tratamiento de la obesidad
Actualmente, la terapia con leptina recombinante humana en pacientes obesos con deficiencia genética de leptina constituye un tratamiento efectivo46. Si bien, ensayos clínicos aleatorizados basados en la administración de diferentes dosis de leptina a pacientes con obesidad común, no han alcanzado los resultados esperados, esto es, una disminución de la ingesta y reducción de su peso corporal47. Datos de estudios recientes sugieren que esta resistencia a la leptina en pacientes con obesidad multifactorial podría deberse a una deficiente señalización intracelular48.
Estudios recientes han puesto de relieve la importancia que ciertas citokinas como el Factor Neurotrófico Ciliar (CTNF) tienen en la regulación de la ingesta y el peso corporal49. Considerando que esta citokina mantiene un importante papel en los procesos de señalización celular y con ello en la regulación de la ingesta y peso corporal, cabe considerar su potencial utilidad como alternativa a los efectos anorexígenos de la leptina. Ello tiene su principal justificación en la idea de evitar con ello los efectos de resistencia propios de la leptina50.
Por último, se ha comprobado cómo intervenciones educativas basadas en la modificación de estilos de vida para reducir peso en pacientes obesos resultan igualmente efectivas en sujetos portadores y no portadores de mutaciones en MC4R51. Considerando todo lo anterior, y dada la importancia del sistema leptina-melanocortinas en la regulación de la ingesta y peso corporal en sujetos obesos, es probable que surjan nuevos tratamientos antiobesidad fundamentados en el uso de agonistas de receptores en este sistema52.
Bibliografía
1. Aguilar Cordero MJ, González Jiménez E, García García CJ, García López PA, Álvarez Ferre J, Padilla López CA et al. Obesity in a school children population from Granada: assessment of the efficacy of an educational intervention. Nutr Hosp 2011; 26: 636-641. [ Links ]
2. Aguilar Cordero MJ, González Jiménez E, Sánchez Perona J, Padilla López CA, Álvarez Ferre J, Mur Villar N et al. The Guadix study of the effects of a Mediterranean-diet breakfast on the postprandial lipid parameters of overweight and obese pre-adolescents. Nutr Hosp 2010; 25: 1025-1033. [ Links ]
3. Ochoa MC, Martí A, Martínez JA. Estudios sobre la obesidad en genes candidatos. Med Clin (Barc) 2004; 122: 542-551. [ Links ]
4. Rankinen T, Zuberi A, Chagnon YC, Weisnagel SJ, Argyropoulos G, Walts B. The human obesity gene map: the 2005 update. Obesity 2006; 14: 529-644. [ Links ]
5. MacNeil DJ, Howard AD, Guan X, Fong TM, Nargund RP, Bednarek MA et al. Eur J Pharmacol 2002; 450: 93-109. [ Links ]
6. Rosadoc EL, Monteiro JB, Chaia V, Do Lago MF. Efecto de la leptina en el tratamiento de la obesidad e influencia de la dieta en la secreción y acción de la hormona. Nutr Hosp 2006; 21: 686-693. [ Links ]
7. Bulló Bonet M. La leptina en la regulación del balance energético. Nutr Hosp 2002; 17 (Supl. 1): 42-48. [ Links ]
8. Robertson SA, Leinninger GM, Myers MG. Molecular and neural mediators of leptin action. Physiol Behav 2008; 94: 637-642. [ Links ]
9. Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci 2005; 8: 571-578. [ Links ]
10. Seeley RJ, Drazen DL, Clegg DJ. The critical role of the melanocortin system in the control of energy intake. Annu Rev Nutr 2004; 24: 133-149. [ Links ]
11. Ivanova E, Kelsey G. Imprinted genes and hypothalamic function. J Mol Endocrinol 2011; 47: 67-74. [ Links ]
12. Hung CC, Luan J, Sims M, Keogh JM, Hall C, Wareham NJ et al. Studies of the SIM1 gene in relation to human obesity and obesity-related traits. Int J Obes (Lond) 2007; 31: 429-434. [ Links ]
13. Holder Jr JL, Butte NF, Zinn AR. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet 2000; 9: 101-108. [ Links ]
14. Ghoussaini M, Stutzmann F, Couturier C, Vatin V, Durand E, Lecoeur C et al. Analysis of the SIM1 contribution to polygenic obesity in the French population. Obesity 2010; 18: 1670-1675. [ Links ]
15. Tolson KP, Gemelli T, Gautron L, Elmquist JK, Zinn AR, Kublaoui BM. Postnatal Sim1 deficiency causes hyperphagic obesity and reduced Mc4r and oxytocin expression. Neurobiol Dis 2010; 30: 3803-3812. [ Links ]
16. Tapia-Arancibia L, Rage F, Givalois L, Arancibia S. Physiology of BDNF: focus on hypothalamic function. Front Neuroendocrinol 2004; 25: 77-107. [ Links ]
17. Gray J, Yeo GS, Cox JJ, Morton J, Adlam ALR, Keogh JM. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes 2006; 55: 3366-3371. [ Links ]
18. Han JC, Liu QR, Jones M. Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N Engl J Med 2008; 359: 918-927. [ Links ]
19. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the Mouse obese gene and its human homologue. Nature 1994; 372: 425-432. [ Links ]
20. Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D. Weight reducing effects of the plasma protein encoded by the obese gene. Science 1995; 269: 543-546. [ Links ]
21. Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997; 387: 903-908. [ Links ]
22. González Jiménez E, Aguilar Cordero MJ, García García CJ, García López PA, Álvarez Ferre J, Padilla López CA. Leptin: a peptide with therapeutic potential in the obese. Endocrinol Nutr 2010; 57: 322-327. [ Links ]
23. Oswal A, Yeo GS. The leptin melanocortin pathway and the control of body weight: lessons from human and murine genetics. Obes Rev 2007; 8: 293-306. [ Links ]
24. Andiran N, Celik N, Andiran F. Homozygosity for two missense mutations in the leptin receptor gene (P316:W646C) in a Turkmenian girl with severe early-onset obesity. J Pediatr Endocrinol Metab 2011; 24: 1043-1045. [ Links ]
25. Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 1998; 391: 900-904. [ Links ]
26. Ren D, Li M, Duan C, Rui L. Identification of SH2-B as a key regulator of leptin sensitivity, energy balance, and body weight in mice. Cell Metab 2005; 2: 95-104. [ Links ]
27. Gantz I, Fong TM. The melanocortin system. Am J Physiol Endocrinol Metab 2003; 284: 468-474. [ Links ]
28. Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 1997; 88: 131-141. [ Links ]
29. Vaisse C, Clement K, Guy-Grand B, Froguel P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet 1998; 20: 113-114. [ Links ]
30. Martí A, Corbalán MS, Forga L, Martínez JA, Hinney A, Hebebrand J. A novel nonsense mutation in the melanocortin-4 receptor associated with obesity in a Spanish population. Int J Obes 2003; 27: 385-388. [ Links ]
31. Brumm H, Mühlhaus J, Bolze F, Scherag S, Hinney A, Hebebrand J et al. Rescue of Melanocortin 4 Receptor (MC4R) Nonsense Mutations by Aminoglycoside-Mediated Read-Through. Obesity (Silver Spring) 2012; 20: 1074-1081. [ Links ]
32. Tao YX. Mutations in the melanocortin-3 receptor (MC3R) gene: Impact on human obesity or adiposity. Curr Opin Investig Drugs 2010; 11: 1092-1096. [ Links ]
33. Bochukova EG, Huang N, Keogh J, Henning E, Purmann C, Blaszczyk K, et al. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature 2010; 463: 666-670. [ Links ]
34. Millington GWM. The role of proopiomelanocortin (POMC) neurones in feeding behaviour. Nutr Metab 2007; 4: 18. [ Links ]
35. Choquet H, Stijnen P, Creemers JW. Genetic and functional characterization of PCSK1. Methods Mol Biol 2011;768: 247-253. [ Links ]
36. Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 2001; 411: 480-484. [ Links ]
37. Lautenbach A, Breitmeier D, Kuhlmann S, Nave H. Human obesity reduces the number of hepatic leptin receptor (ob-R) expressing NK cells. Endocr Res 2011; 36:158-166. [ Links ]
38. Wauman J, Tavernier J. Leptin receptor signaling: pathways to leptin resistance. Front Biosci 2011; 1: 2771-2793. [ Links ]
39. Myers MG. Keeping the fat off with nesfatin. Nat Med 2006; 12: 1248-1249. [ Links ]
40. Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci 2003; 6: 736-742. [ Links ]
41. Hu Y, Russek SJ. BDNF and the diseased nervous system: a delicate balance between adaptive and pathological processes of gene regulation. J Neurochem 2008; 105: 1-17. [ Links ]
42. Han JC, Liu QR, Jones M, Levinn RL, Menzie CM, Jefferson-George KS, et al. Brain derived neurotrophic factor and obesity in the WAGR syndrome. N Engl J Med 2008; 359: 918-927. [ Links ]
43. Yeo GS, Connie Hung CC, Rochford J, Keogh J, Gray J, Sivaramakrishnan S. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci 2004; 7: 1187-1189. [ Links ]
44. Gray J, Yeo GS, Cox JJ, Morton J, Adlam ALR, Keogh JM et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes 2006; 55: 3366-3371. [ Links ]
45. Noble EE, Billington C, Kotz CM, Wang C. The lighter side of BDNF. Am J Physiol 2011; 300: 1053-1069. [ Links ]
46. Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med 1999; 341: 879-884. [ Links ]
47. Donato J Jr, Cravo RM, Frazao R, Elias CF. Hypothalamic sites of leptin action linking metabolism and reproduction. Neuroendocrinology 2011; 93: 9-18. [ Links ]
48. Dhillon SS, McFadden SA, Chalmers JA, Centeno ML, Kim GL, Belsham DD. Cellular leptin resistance impairs the leptin-mediated suppression of neuropeptide y secretion in hypothalamic neurons. Endocrinology 2011; 152: 4138-4147. [ Links ]
49. Schuster B, Kovaleva M, Sun Y, Regenhard P, Matthews V, Grötzinger J et al. Signaling of human ciliary neurotrophic factor (CNTF) revisited. The interleukin-6 receptor can serve as an alpha-receptor for CTNF. J Biol Chem 2003; 278: 9528-9535. [ Links ]
50. Reinehr T, Hebebrand J, Friedel S, Toschke AM, Brumm H, Biebermann et al. Lifestyle intervention in obese children with variations in the melanocortin 4 receptor gene. Obesity 2009; 17: 382-389. [ Links ]
51. Beckers S, Zegers D, Van Gaal LF, Van Hul W. The role of the leptin-melanocortin signalling pathway in the control of food intake. Crit Rev Eukaryot Gene Expr 2009; 19: 267-287. [ Links ]
52. González Jiménez E. Genes and obesity: a cause and effect relationship. Endocrinol Nutr 2011; 58: 492-496. [ Links ]
53. Zurbano Inchusta R, Ochoa Nieto MC, Moreno-Aliaga MJ, Marti del Moral A. Estudios sobre obesidad de origen monogénico en humanos. Rev Esp Obes 2004; 2: 269-278. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Dr. Emilio González Jiménez
Departamento de Enfermería
Facultad de Enfermería (Campus de Melilla)
Universidad de Granada
C/ Santander, 1
52071-Melilla (España)
E-mail: emigoji@ugr.es
Recepción: 30 de noviembre de 2011
Aceptación provisional: 7 de marzo de 2012
Aceptación definitiva: 16 de marzo de 2012

