










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Clínica de Medicina de Familia
versión On-line ISSN 2386-8201versión impresa ISSN 1699-695X
Rev Clin Med Fam vol.9 no.3 Barcelona oct. 2016
Poliquistosis renal
Polycystic kidney disease
Miguel Ángel Babiano Fernández y Alfredo Rodríguez Fernández
Médico de Familia. Centro de Salud de Argamasilla de Calatrava. Argamasilla de Calatrava Ciudad Real. (España).
El presente trabajo fue presentado como comunicación en las III Jornadas Nacionales Nefrourológicas de SEMERGEN (Granada, 24-26 de abril de 2015).
Dirección para correspondencia
RESUMEN
Presentamos el caso de un varón de 45 años con poliquistosis renal dominante, al que desde hace años se le realiza el seguimiento de su patología renal desde Atención Primaria.
La poliquistosis renal es una enfermedad hereditaria que puede tener un patrón de transmisión autosómica dominante o recesiva, manifestándose clínicamente este último desde las primeras etapas de la vida.
Se trata de una enfermedad poco frecuente y desconocida para muchos médicos, por lo que es esencial conocer su existencia con la finalidad de realizar un diagnóstico precoz y evitar la progresión general de la enfermedad hacia la insuficiencia renal.
En Atención Primaria, nuestro objetivo prioritario será el control de la hipertensión arterial y de las infecciones urinarias, para evitar la progresión de la enfermedad, así como la búsqueda activa de familiares afectos.
El uso de la ecografía en Atención Primaria nos permite realizar tanto el diagnóstico como el seguimiento, así como el cribado de familiares de pacientes con la enfermedad.
Palabras clave: Enfermedad Renal. Riñón Poliquístico Autosómico Recesivo. Riñón Poliquístico Autosómico Dominante.
ABSTRACT
We report the case of a 45-year-old male with dominant polycystic kidney disease who has received follow-up in Primary Care for years.
Polycystic kidney disease is an inherited disease that can have an autosomal dominant or recessive pattern of transmission, being the latter clinically manifest from the earliest stages of life. This disease is rare and unknown to many physicians. It is therefore essential to be aware of its existence in order to make an early diagnosis and prevent the overall progression of the disease towards kidney failure.
At primary care level, our priority objective will be the control of arterial hypertension and urinary tract infections to prevent the progression of the disease, as well as the active search for affected relatives.
The use of ultrasound in primary care allows us to perform both diagnosis and monitoring of our patient, and to perform screening of the patient's relatives with the disease.
Key words: Kidney Disease. Polycystic Kidney, Autosomal Recessive. Polycystic Kidney, Autosomal Dominant.
Introducción
La poliquistosis renal dominante del adulto es la enfermedad hereditaria más frecuente en la población española, con una frecuencia aproximada de 1/1.000 individuos1.
Al ser los síntomas iniciales de la enfermedad inespecíficos, se retrasa su diagnóstico hasta etapas avanzadas de la enfermedad, siendo fundamental la sospecha diagnóstica en las fases iniciales de la misma.
También debemos evitar la progresión de la enfermedad hacia la insuficiencia renal avanzada mediante el control estricto de la tensión arterial y de la infección renal.
En atención primaria disponemos de la ecografía abdominal, que es la mejor prueba de imagen disponible para el diagnóstico y seguimiento de estos pacientes, así como para realizar el cribado de familiares de pacientes con enfermedad conocida.
Caso clínico
Varón de 45 años con antecedentes personales de hepatitis C crónica, hipertensión arterial (HTA) y enfermedad renal crónica (ERC) en estadio V, secundaria a poliquistosis renal de más de 10 años de evolución, que fue diagnosticada en el servicio de Digestivo, donde se le seguía desde hacía años por la hepatitis C que padecía. En la actualidad, el paciente se encuentra en programa de diálisis.
Respecto a su familia, su madre tiene una poliquistosis renal, y tiene un hijo sano, al que se le realizan controles ecográficos para la detección precoz de la enfermedad.
La exploración física en el momento actual es anodina, con peso y talla de 68 Kg y 172 cm respectivamente, IMC de 22,98, y controles de presión arterial en torno a 130/70 mmHg. Llama la atención que, al tratarse de un paciente delgado, la simple palpación abdominal permite apreciar varias masas abdominales en ambas fosas renales.
La ecografía abdominal realizada en el centro de salud muestra múltiples quistes renales de carácter bilateral de contenido hipoecoico con refuerzo posterior sin apenas parénquima renal sano (figuras 1 y 2).
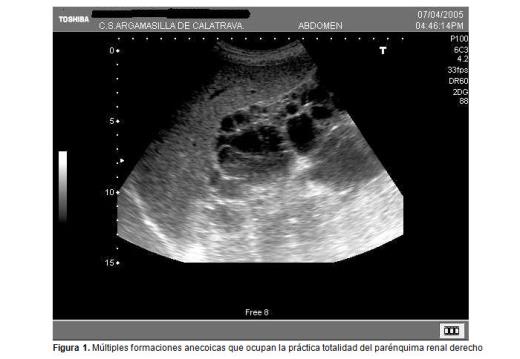
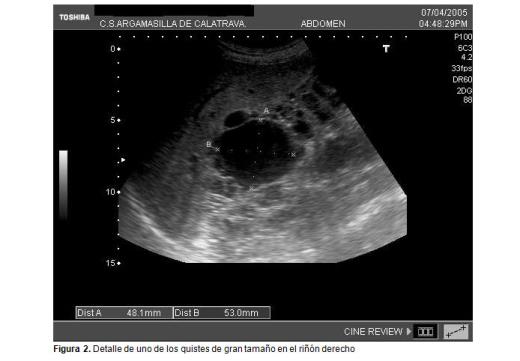
En la actualidad, en la consulta de atención primaria se le realiza seguimiento de cumplimiento terapéutico, pues está en situación de diálisis, y controles ecográficos a su hijo, que hasta ahora han sido normales.
Discusión
La poliquistosis renal es la enfermedad renal hereditaria más frecuente. Su prevalencia estimada es de 1 cada 800-1.000 personas. Se trata de una enfermedad hereditaria que puede ser de transmisión autosómica dominante o recesiva, esta última con manifestaciones clínicas desde las primeras etapas de la vida.
Los pacientes con poliquistosis renal autosómica dominante (PRAD) constituyen aproximadamente un 8-10 % de la población en diálisis1, suponiendo la tercera causa de insuficiencia renal terminal. La hipertensión arterial es la manifestación clínica más frecuente2, siendo la poliquistosis una de las causas de hipertensión secundaria y el principal factor que contribuye a la progresión de la enfermedad3,4.
La variante autosómica dominante de la poliquistosis renal es una de las enfermedades hereditarias más frecuentes. Afecta a una de cada mil personas y se caracteriza por el desarrollo progresivo de quistes renales5. En la actualidad se estudian factores de crecimiento que podrían ser los responsables del desarrollo anómalo del epitelio tubular; es por ello que muchos de estos pacientes presentan también quistes en hígado, páncreas y bazo, habitualmente bien tolerados y que no causan ninguna sintomatología. Existen dos genes causantes: el gen PKD1, localizado en el cromosoma 16; y el gen PKD2, localizado en el cromosoma 4. El 85 % de las poliquistosis renales autosómicas dominantes son causadas por el gen PKD1, y el resto por mutaciones en el gen PKD2. La enfermedad asociada al gen PKD1 es más grave que la asociada al gen PKD2, con inicio en edades tempranas6.
Como todas las enfermedades con herencia autosómica dominante, la presencia de una sola copia del gen mutado es suficiente para que la enfermedad se manifieste. Las principales características de este patrón de herencia son la transmisión vertical (en la que cada paciente tiene al padre o a la madre afectados por la enfermedad), el mismo riesgo para ambos sexos de padecer o transmitir la enfermedad y una probabilidad del 50 % de que los descendientes estén afectados.
Las manifestaciones de la enfermedad pueden surgir en la tercera o cuarta década de la vida. Generalmente, los síntomas iniciales de la poliquistosis renal son inespecíficos y difíciles de detectar, por lo que el papel del médico de atención primaria en las fases iniciales es clave para su diagnóstico.
Debemos sospechar una poliquistosis renal en todo paciente con antecedentes familiares de patología crónica renal que presente un cuadro de dolor lumbar o abdominal en presencia de masa, hematuria7, hipertensión arterial o insuficiencia renal.
La mayoría de estos pacientes, cuando son diagnosticados, se encuentran en fases avanzadas y requieren un tratamiento especializado por parte del nefrólogo. El único objetivo del tratamiento es disminuir la progresión de la enfermedad mediante el control de la hipertensión arterial y de la insuficiencia renal.
Actualmente, existen estudios en animales con antagonistas de los receptores V2 de la vasopresina que parecen disminuir el crecimiento de los quistes.
Aproximadamente, la mitad de estos pacientes progresan rápidamente hacia la insuficiencia renal terminal necesitando, como última alternativa terapéutica, la diálisis y el trasplante renal. Aproximadamente el 10 % de los enfermos sometidos a diálisis presentan esta patología.
Se trata de una enfermedad poco frecuente y desconocida para muchos médicos, por lo que es esencial conocer su existencia con la finalidad de realizar un diagnóstico precoz y de conocer las implicaciones que la enfermedad supone para el paciente. El diagnóstico precoz de la PRAD conllevaría un mejor pronóstico, al permitir un seguimiento clínico más estricto, cumpliendo los objetivos terapéuticos de forma temprana y de esa forma retrasando el avance de la enfermedad renal. La hipertensión y la infección renal se deben combatir de manera intensiva apenas se tenga el diagnóstico, para preservar la función renal.
El mayor beneficio es la posibilidad de planificación familiar, detección y tratamiento temprano de las complicaciones de la enfermedad.
Es de destacar en atención primaria el conocimiento de la familia para que en determinados procesos de carácter hereditario, como es este caso, podamos intervenir realizando un diagnóstico precoz. Para ello se dispone de la ecografía abdominal como la mejor modalidad de imagen8, tanto para el diagnóstico como para hacer el cribado en familias de pacientes afectados, así como en el seguimiento sistemático de pacientes que tienen enfermedad conocida.
Bibliografía
1. Marrero Robayna S, Hortal Cascón L, Vega Díaz N, Rodríguez Pérez JC. Nefropatías hereditarias y congénitas. Medicine. 2015; 11 (80): 4793-802. [ Links ]
2. Schrier RW, Abebe KZ, Perrone RD, Torres VE, Braun WE, Steinman TI, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med. 2014; 371 (24): 2255-66. [ Links ]
3. Torres-Sánchez MJ, Ávila-Barranco E, Esteban de la Rosa RJ, Fernández-Castillo R, Esteban MA, Carrero JJ, et al. Relación entre función y volumen renal en la poliquistosis renal autosómica dominante: estudio transversal. Rev Clin Esp. 2016; 216 (2): 62-7. [ Links ]
4. Sebastián Aparicio MP, Quintana Urrea E, Garay López de Aguileta AM. Poliquistosis renal causa secundaria de hipertensión. FMC. 2014; 21 (3): 195-6. [ Links ]
5. Gabow PA. Autosomal dominant polycystic kidney disease. N Eng J Med. 1993; 329 (5): 332-42. [ Links ]
6. Ariza M, Álvarez V, Sanz de Castro S, Peces R, Aguado S, Álvarez J, et al. Análisis mutacional del gen PKD1 en pacientes con poliquistosis renal dominante. Nefrología. 1998; XVIII (5): 382-8. [ Links ]
7. Sevillano AM, Gutiérrez E. Protocolo diagnóstico de la microhematuria. Medicine. 2015; 11 (82): 4924-6. [ Links ]
8. Rumack CM, Wilson SR, Charboneau JW, Johnson J-AM. Diagnóstico por ecografía. 3a ed. Madrid: Elsevier; 2006; p.372-3. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Miguel Ángel Babiano Fernández.
Centro de Salud de Argamasilla de Calatrava.
C/ Pinto, 14. C.P. 13440
Argamasilla de Calatrava (Ciudad Real).
España.
Correo electrónico: mababiano@sescam.jccm.es.
Recibido el 29 de noviembre de 2015.
Aceptado para su publicación el 20 de mayo de 2016.

