










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCCIÓN
El linfoma de tejido linfoide asociado a mucosas (MALT) es una entidad poco común de linfoma no Hodgkin (LNH), que se desarrolla a expensas de las células B ubicadas en el tejido linfoide de las membranas mucosas. Su localización en las glándulas salivales es infrecuente, representando el 4% de todos los linfomas. Entre los linfomas parotídeos, el 20% corresponde a linfomas tipo Hodgkin (LH) y el 80% a LNH. Dentro de este último tipo, se encuentran los linfomas MALT, también conocidos como linfomas de células B monocitoides, siendo el subtipo más común de linfoma extranodal primario y, por ende, el más frecuentemente encontrado en la glándula parótida [1-2]. Los linfomas de glándulas salivales pueden presentar un retraso en su diagnóstico debido a su comportamiento relativamente benigno, que mimetiza a tumores más frecuentes del área parotídea.
El objetivo de este estudio es realizar una revisión actualizada de la epidemiología, clínica, diagnóstico, tratamiento y pronóstico de los tumores MALT de parótida.
MATERIAL Y MÉTODO
Búsqueda bibliográfica en PubMed, EMBASE y WoS. Los términos utilizados en la búsqueda fueron lymphoma MALT, parotid gland y monocytoid B-cell lymphoma. Como criterios de selección, se incluyeron todos los estudios clínicos y epidemiológicos en humanos, descartando los estudios en animales y los centrados en el ámbito inmunohistoquímico y de genética molecular, los artículos que no estuvieran en inglés o español y los estudios con material redundante. Se abarcaron todos los estudios de casos y series de casos de cualquier edad y sexo con diagnóstico de linfoma MALT de glándula parótida, que incluyeran datos de clínica, diagnóstico y tratamiento. Dado el limitado número de estudios encontrados, no se aplicaron filtros adicionales y se procedió a una selección manual para el estudio cualitativo. La revisión se realizó siguiendo los criterios PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-analysis) [3].
RESULTADOS
En total, se seleccionaron 28 estudios originales de casos o series que cumplían los criterios de inclusión, de los que 10 de ellos incluyeron más de 25 pacientes. En la Figura 1 se resume el proceso de selección de los artículos. La Tabla 1 recoge los artículos seleccionados.
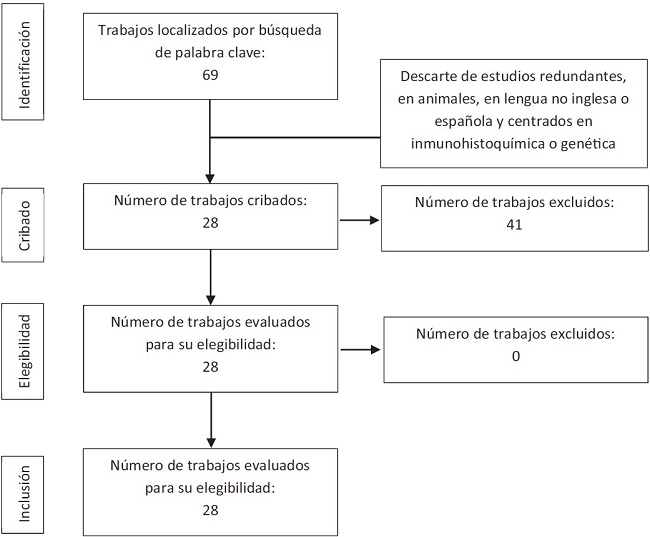
Tabla 1. Linfoma MALT de glándula parótida. Artículos incluidos en la revisión.
|
Autor |
Tipo de Estudio |
País |
Casos |
Objetivo de estudio |
|---|---|---|---|---|
|
Zhang et al. 2021 |
Retrospectivo, multicéntrico |
China |
72 |
Etiopatogenia |
|
Mantsopoulo et al. 2021 |
Retrospectivo, multinacional |
Grecia Alemania |
67 |
Diagnóstico por imagen |
|
Beckers et al. 2020 |
Caso clínico |
México |
1 |
Descripción y revisión |
|
Toni et al. 2020 |
Caso clínico |
Italia |
1 |
Descripción y revisión |
|
Hwang et al. 2019 |
Caso clínico |
Corea |
1 |
Descripción y revisión |
|
Aydin et al. 2016 |
Caso clínico |
India |
1 |
Descripción y revisión |
|
Bennett et al. 2015 |
Caso clínico |
EE. UU. |
1 |
Descripción y revisión |
|
Vázquez et al. 2015 |
Retrospectivo multicéntrico |
EE. UU. |
507 |
Epidemiología y supervivencia |
|
Jackson et al. 2015 |
Retrospectivo multicéntrico |
Internacional |
247 |
Tratamiento y pronóstico |
|
Shum et al. 2014 |
Caso clínico |
EE. UU. |
1 |
Descripción y revisión |
|
Zhu et al. 2013 |
Retrospectivo, unicéntrico. |
China |
34 |
Diagnóstico por imagen |
|
Anacak et al. 2012 |
Retrospectivo multicéntrico |
EE. UU. |
63 |
Clínica, tratamiento, pronóstico |
|
Fernández et al. 2012 |
Caso clínico |
Chile |
1 |
Descripción y revisión |
|
Pollard et al. 2011 |
Restrospectivo unicéntrico |
EE. UU. |
329 |
Asociación con SS, tratamiento |
|
Troch et al. 2010 |
Retrospectivo unicéntrico |
EE. UU |
24 |
Pronóstico |
|
Frómeta et al. 2010 |
Caso clínico |
Cuba |
1 |
Descripción y revisión |
|
Pino et al. 2007 |
Caso clínico |
Portugal |
1 |
Descripción y revisión |
|
Tada et al. 2005 |
Caso clínico |
Japón |
1 |
Descripción y revisión |
|
Ando M et al. 2005 |
Caso clínico |
Japón |
1 |
Descripción y revisión |
|
Vaíllo et al. 2004 |
Caso clínico |
España |
1 |
Descripción y revisión |
|
Marioni et al. 2004 |
Caso clínico |
Italia |
1 |
Descripción y revisión |
|
Tsang et al. 2003 |
Retrospectivo multicéntrico |
EE. UU. |
103 |
Tratamiento |
|
Rosenstiel et al. 2001 |
Caso clínico |
EE. UU. |
1 |
Descripción y revisión |
|
Biasi et al. 2001 |
Caso clínico |
Italia |
6 |
Descripción y revisión |
|
Zinzani et al. 1999 |
Retrospectivo, multicéntrico |
Italia |
75 |
Comportamiento según localización |
|
Damborenea et al.1998 |
Caso clínico |
España |
1 |
Descripción y revisión |
|
Thieblemont et al. 1997 |
Retrospectivo, multicéntrico |
EE. UU. |
108 |
Descripción y revisión |
|
Balm et al. 1993 |
Retrospectivo, unicéntrico |
Holanda |
7 |
Tratamiento |
SS = Síndrome de Sjögren.
Vázquez et al [4] estudiaron retrospectivamente 507 casos de linfoma MALT de glándulas salivales, de los cuales 370 (73%) se produjeron en mujeres, 374 (75%) en >50 años, la glándula parótida estuvo implicada en el 81% de los mismos y 273 pacientes (54%) presentaron un estadio I en el momento del diagnóstico. No encontraron diferencias en el pronóstico asociadas al tipo de tratamiento (cirugía y/o RT), pero sí asociadas a la raza (peor pronóstico en afroamericanos) y al estadio (peor pronóstico en los estadios más avanzados).
Otro estudio retrospectivo de 13 centros pertenecientes a la red de Cánceres Raros (EURACAN) de 10 países distintos incluyó 63 pacientes con linfoma MALT de glándulas salivales. La edad media fue de 58 años, y 47 pacientes fueron mujeres. La glándula parótida estuvo implicada en 49 casos, y en 9 pacientes hubo varias glándulas implicadas. La mayoría se detectaron en estadio IE. La tasa de recurrencia a los 5 años fue del 35%. Sin embargo, ésta no afectó a la supervivencia. En esta serie, sólo la radioterapia (RT) aumentó la supervivencia libre de enfermedad [5].
Otro estudio relevante de linfoma MALT de parótida, incluyó 28 casos retrospectivos, todos los tratados en el Hospital Universitario de Viena durante 11 años [6]. En el estudio, se evidencian 19 (68%) mujeres, con una media de edad de 49 años. En 10 (36%) casos se presentó enfermedad con diseminación a distancia (25% con afectación parotídea bilateral y el resto con afectación de otras zonas extranodales). Se evidenció enfermedad autoinmune subyacente en 18 (64%) pacientes, siendo el síndrome de Sjögren la entidad más frecuente (78%). No se observaron diferencias en la evolución clínica entre los casos con enfermedad autoinmune y los libres de la misma. En 12 (46%) casos se presentó alguna recaída (media de tiempo de recaída: 18 meses). No se observó asociación con diseminación a tracto gastro-intestinal. La supervivencia a los 5 años fue del 86%, no obstante, solo un paciente falleció por recaída del linfoma.
Mantsopoulos et al [7] examinaron retrospectivamente una serie de 66 pacientes diagnosticados entre 2005 y 2017de linfoma de glándula parotídea. Las entidades histológicas más frecuentes fueron linfoma MALT (31%) y linfoma folicular (28%). El estadio I es el más frecuente al diagnóstico (52%). El diagnóstico se realizó tras parotidectomía en 50 (76%) casos, mediante biopsia de aguja fina en 14 (21%) y biopsia abierta en el resto de los pacientes (3%). Se destacó que los linfomas de glándula parótida pueden fácilmente mimetizar tanto condiciones benignas como malignas y que deben incluirse siempre en el diagnóstico diferencial de prácticamente todos los tipos de lesiones parotídeas.
Zhang et al [8] estudiaron una serie retrospectiva de 72 casos del hospital Shanghai Ninth People's. La relación varón/mujer fue 1:2,8, y la edad mediana de 57 años. En 9 (12,5%) pacientes se evidenciaron múltiples masas. Entre los que presentaron una masa única, la glándula parótida estuvo implicada en 54 (75%) individuos. Se presentó una enfermedad autoinmune en 25 (35%) de casos, siendo el síndrome de Sjögren el más frecuente. La trisomía 3 constituyó el cambio epigenético más frecuente (58%), especialmente en pacientes sin síndrome de Sjögren, seguida de la trisomía 18 (10%). La supervivencia a los 5 años fue del 94% y la supervivencia libre de enfermedad a los 5 años fue del 85%. La trisomía 18 presentó mayor posibilidad de recurrencia.
DISCUSIÓN
Los linfomas abarcan un heterogéneo grupo de tumores que nacen de los sistemas reticuloendotelial y linfático y se originan principalmente en los nódulos linfáticos. Los tipos principales son el linfoma Hodgkin y linfoma no Hodgkin. Los LNH corresponden al 25% de todos los cánceres de cabeza y cuello y se incluyen en menos del 5% de los tumores malignos de la región parotídea [9]. Los linfomas MALT, descritos inicialmente en 1983, son un subtipo de LNH constituido por grupos no encapsulados de linfocitos localizados en el tejido mucoso del tracto aéreo-digestivo [10].
Los tumores de la glándula parótida representan el 6-8% de todos los tumores de cabeza y cuello [11]. Los tumores benignos de la glándula parótida constituyen la gran mayoría de los tumores de las glándulas salivales. Entre los linfomas parotídeos, el 80% corresponde a LNH, y los más frecuentes son el tipo folicular, difuso de células grandes y asociado a mucosas (MALT) [12]. Este último representa alrededor del 5% de los LNH diagnosticados; sin embrago, es el subtipo más común de linfoma extranodal [13]. Su incidencia anual se estima en alrededor de 1/313000 [14]. Afecta mayoritariamente a personas mayores de 60 años, con predominio en mujeres sobre varones, en una proporción 1,5:1, en probable relación con la mayor afectación de enfermedades autoinmunes en las mismas. Es muy poco frecuente en niños [14].
El linfoma MALT afecta principalmente al tracto gastrointestinal (más del 70%), aunque puede abarcar potencialmente cualquier localización que albergue linfocitos B, describiéndose en glándulas salivales, tiroides, pulmón, anexos oculares, timo, mama o riñón [15]. El área de cabeza y cuello representa el 34% de los casos extraintestinales, localizándose, en orden de frecuencia, en el anillo de Waldeyer, senos paranasales y glándulas salivales [16]. Dentro de este último grupo, las glándulas más afectadas son la parótida, con el 75% de los casos, seguida de la submaxilar (23%) y la sublingual (1%) [17].
Los tumores de la glándula parotídea se relacionan principalmente con radiación [18], tabaco y ciertas infecciones virales (Epstein-Barr, HIV, VPH) [11]. Hasta en el 44% de los pacientes con linfoma MALT se asocian enfermedades autoinmunes, especialmente síndrome de Sjögren [19-26]. Este síndrome ataca a las glándulas exocrinas, especialmente a las glándulas salivales, y da lugar a una proliferación significativa de células B [27], la cual representa un importante factor de riesgo para el desarrollo de un linfoma de glándulas salivales. El incremento del riesgo puede ser de hasta 10 veces superior en un paciente con síndrome de Sjögren [28]. Por lo tanto, el linfoma MALT de glándula parótida debe tenerse en cuenta en el diagnóstico diferencial de cualquier paciente con síndrome de Sjögren que presenta clínica de neoplasia de glándula parótida [29], incluso en niños [30]. Pese a la relación de enfermedades autoinmunes de base con la aparición de linfomas, su presencia no parece afectar al pronóstico del linfoma MALT. La íntima asociación entre el linfoma tipo MALT y condiciones infecciosas crónicas o enfermedades autoinmunes ha sido bien establecida en el caso de infección por Helicobacter pylori y su localización en el estómago [1, 2]. No obstante, su etiología ha sido menos estudiada y establecida en apariciones extraestomacales.
Los LNH de la glándula parotídea se presentan habitualmente como una masa indolente de crecimiento lento. Otros síntomas menos frecuentes incluyen inflamación parotídea bilateral, linfadenopatía cervical, dolor o paresia del nervio facial. La presentación clínica es indistinguible de otras inflamaciones de la glándula y el diagnóstico diferencial incluye tumores benignos y malignos de la glándula parótida.
Antes de proceder a los estudios de imagen, se debe obtener una detallada historia clínica y realizar una exploración física exhaustiva para decidir la técnica de imagen más efectiva a utilizar. Los detalles de la anamnesis incluyen tiempo de evolución de la masa, ritmo de crecimiento, presencia de dolor o molestias faciales, historia previa de cáncer de cabeza, cuello o piel, y presencia de comorbilidades (por ejemplo, enfermedades autoinmunes). La exploración física debe detallar tamaño, movilidad, fijación de la masa a estructuras anatómicas subyacentes, presencia de linfadenopatías locales o distantes y hallazgos sugestivos de parálisis del nervio facial [31, 32].
La evaluación diagnóstica inicial de una masa parotídea debe incluir TAC y RMN, para determinar la localización, forma y tamaño, así como la presencia de afectación extraglandular, linfadenopatía cervical y relación con otras estructuras vitales. Hallazgos óseos como erosión o expansión del hueso temporal o mandibular se aprecian mejor con TAC, mientras que la infiltración de tejidos blandos, extensión intracraneal e invasión perineural se detectan mejor con RMN [31, 33]. Considerando la localización superficial de las glándulas salivales, las imágenes por ultrasonidos (US) pueden jugar un papel en el diagnóstico por su rapidez y coste-efectividad [31].
No se han reportado hallazgos específicos en las imágenes radiológicas en el linfoma MALT de glándula parótida, y pueden encontrarse también en otros trastornos linfoproliferativos, por ejemplo, en pacientes con síndrome de Sjögren y LESA. Los US y el TAC muestran frecuentemente una lesión localizada o difusa en la glándula acompañada de múltiples quistes, que probablemente representan dilataciones focales de los ductos salivales producidos por la compresión de conductos terminales por los linfocitos neoplásicos. Estos quistes pueden asociar calcificaciones, probablemente resultado de lesiones inflamatorias [34]. Raramente, se pueden detectar granulomas [35]. No se suele detectar linfadenopatía cervical evidente [36]. El 50% de los casos presentan afectación multifocal sincrónica. Se ha descrito afectación parotídea bilateral [37, 38] y coexistencia de tumor de Warthin y MALT [45], especulándose que un estímulo inmunológico a largo plazo por el tumor de Warthin podría causar un tumor MALT de parótida.
Ocasionalmente se ha descrito la presentación del linfoma MALT como una única masa quística de glándula salival [39]. Por tanto, el diagnóstico de un quiste linfoepitelial ha de incluir la posibilidad de un linfoma MALT concurrente, especialmente en el caso de condiciones de riesgo como una linfocitosis monoclonal de células B [40]. En estos casos, debe realizarse una resección quirúrgica completa para establecer el diagnóstico [41].
En cuanto al papel del PET, detecta el 67% de los linfomas MALT de la zona marginal y constituye una prueba fundamental para detectar formas multifocales y conocer su extensión, así como para definir el plan de tratamiento, determinar el pronóstico y permitir comparaciones durante el seguimiento [31].
El diagnóstico definitivo de los linfomas MALT se basa en la histología del la lesión, hematología y análisis bioquímicos. Para ello, además de los estudios de imagen, se obtiene rutinariamente un FNAB o PAAF (biopsia por aspiración con aguja fina), para análisis citológico. El uso de la PAAF en linfomas de cabeza y cuello es seguro [42, 43], pero presenta una tasa del 32% de falsos negativos. Además, su habilidad para diferenciar el tipo histológico de linfoma es baja. Por ello, debido a la dificultad para obtener una muestra definitoria mediante FNAB o PAAF, frecuentemente se debe realizar una parotidectomía parcial para el diagnóstico patológico [31].
Histológicamente, los linfocitos de los linfomas MALT son células marginales de tamaño pequeño o mediano y se clasifican como linfomas extranodales de células marginales B [44]. Ocasionalmente pueden presentarse asociados a células plasmáticas y macrófagos [45]. Aunque la mayoría de los linfomas de parótida se describen como LNH extranodales, algunos pueden provenir del tejido linfoide asociado a la glándula parótida. El parénquima glandular se suele afectar secundariamente, siendo difícil establecer el origen del linfoma. A pesar de esta controversia, el tipo de tratamiento no cambia.
Debido a la escasez de casos descritos, el tratamiento de los linfomas MALT es controvertido. Generalmente se describe el empleo de radioterapia localizada para estadios I y II, mientras que la terapia sistémica se reserva para estadios avanzados.
El linfoma MALT se considera muy sensible a la RT. El régimen más empleado para estadios iniciales es en dosis de 24 Gy repartida en 12 fracciones [46, 47]. En un estudio retrospectivo de 247 pacientes con linfoma MALT en glándulas salivales, dirigido por Jackson et al [48], la terapia inicial consistió en RT y/o cirugía (57%), quimioterapia (QT) sistémica y/o rituximab (37%) y observación (6%). La media de supervivencia global fue de 18,3 años y no se observaron diferencias entre los pacientes que recibieron tratamiento local o sistémico. Tsang et al [49] estudiaron 103 pacientes en estadio I y II de linfoma MALT y observaron que la radioterapia a dosis moderadas (25-35 Gy) permitía un control local excelente. Por otro lado, otros estudios sugieren que, en los linfomas MALT de parótida de bajo grado, la quimioterapia sola presenta resultados similares a la RT aislada [24, 41].
Considerando una mayor morbilidad de la terapia sistémica, se puede establecer que la quimioterapia no parece necesaria en fases iniciales. Sin embargo, en subtipos más agresivos, como en linfomas de células B grandes difuso, se suele recomendar quimioterapia más RT [50]. El agente más utilizado es el rituximab a dosis estándar (375mg/m2), administrado semanalmente durante un mes. Logvinenko trató con rituximab a 17 pacientes con linfoma MALT asociado a síndrome de Sjögren y reportó la eliminación de signos histológicos de MALT en el 71% de los casos tras 6 meses de tratamiento [51]. Por otra parte, Pollard et al destacó, en una serie retrospectiva de MALT asociado a Sjögren, que la observación expectante en casos de linfoma MALT asintomático en ausencia de síndrome de Sjögren activo, puede ser una opción adecuada pues la mayoría de estos pacientes permanecieron asintomáticos durante un periodo de tiempo prolongado (2-12 años observados, mediana: 6 años) [52].
Se recomienda quimioterapia sin radioterapia asociada en las formas avanzadas o tras una resección incompleta del tumor. Por otra parte, dado que los pacientes con LESA tienen un riesgo 44 veces mayor de desarrollar linfoma maligno de glándulas salivales o extrasalivales, de los cuales el 80% son de tipo MALT [11], se requiere un seguimiento estrecho con nueva biopsia de cualquier lesión persistente o recurrente en este tipo de pacientes [41].
Mientras que la cirugía juega un papel considerable en el manejo de la mayoría de las neoplasias de la glándula parótida, se considera innecesariamente invasiva en el tratamiento de los linfomas de glándula parótida [31]. Su papel queda reservado para tumores de localización única en pacientes con contraindicación a la RT, aunque estos casos son excepcionales debido a las bajas dosis de radiación requeridas [31, 53]. No se han encontrado estudios que establezcan técnicas quirúrgicas de elección, describiéndose exéresis glandulares parciales con preservación del nervio facial. Existen autores que describen mayor riesgo de diseminación solo con tratamiento quirúrgico y recomiendan asociar siempre quimioterapia y/o radioterapia [41].
El curso clínico de los linfomas MALT difiere del de otros linfomas de bajo grado. La mayoría de los estudios previos han mostrado evoluciones favorables [20, 49, 54-56]. Aproximadamente el 70% de los pacientes con linfoma MALT se presentan en un estadio I o II de la enfermedad [41]. En cuanto al pronóstico, los linfomas MALT de parótida, en comparación con otras localizaciones extranodales de LNH, suelen ser más frecuentemente de bajo grado y presentan mejor pronóstico, con una supervivencia mayor al 80% a los 5 años [4, 31].
Thieblemont et al [55] estudiaron a 108 pacientes con linfoma MALT y observó que los pacientes con afectación no gastrointestinal (53 pacientes) parecen progresar más frecuentemente que los que presentan afectación gastrointestinal. A pesar de que varios autores [6, 56-57] demostraron el pronóstico favorable de pacientes con linfomas MALT extraintestinales, éstos presentaron mayor tasa de recaídas, incluyendo en las glándulas salivales, que los pacientes con linfomas MALT gastrointestinales o tiroideos. Tonami et observó 3 casos de linfoma MALT de glándulas salivales con síndrome de Sjögren que progresaron a otras zonas mucosas, a pesar de sufrir una remisión completa en la localización primaria al [21]. Harris comentó que la razón principal para tratar los linfomas MALT de glándula parótida es la prevención del desarrollo de linfomas de alto grado [16].
CONCLUSIONES
Los linfomas MALT de glándulas salivales son una entidad infrecuente. Su localización más habitual es la glándula parótida y su presentación indolente se mimetiza con otras tumoraciones benignas más comunes. Su diagnóstico se realiza mediante el hallazgo de lesiones quísticas difusas en el TAC y RMN. Es importante tener en cuenta la tasa alta de falsos negativos en la PAAF, por lo que nunca podremos descartar su sospecha ante resultados inespecíficos. En ocasiones, se requiere realizar parotidectomía para establecer el diagnóstico definitivo. Su tratamiento incluye la cirugía, radioterapia y/o quimioterapia, según su localización o subtipo histológico. La tasa de supervivencia es elevada.

