










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroducción
Este artículo especial es la repuesta a una convocatoria que JONNPR realizó en 2017 a raíz del premio Nobel de Fisiología y Medicina concedido a tres americanos, Jeffrey C. Hall, Michael Rosbash y Michael W. Young por sus descubrimientos sobre los mecanismos moleculares que controlan el ritmo circadiano(1). En aquella publicación surgió la invitación a nuestros lectores para colaborar en artículos que revisaran y trataran cualquier tema premiado con el Nobel entre los años 2000 y 2016(1). Posteriormente también hemos hecho la misma convocatoria(2-5). Hace unos meses, publicábamos una editorial sobre el Premio Nobel de Fisiología o Medicina de 2021(5) que premiaba la labor y trayectoria de David Julius, fisiólogo estadounidense de 66 años y Ardem Patapoutian, biólogo estadounidense de origen libanés de 54 años, por sus descubrimientos sobre los receptores TRP relacionados con el tacto, temperatura y la presión dada su importancia decisiva para la salud(5). En ella señalábamos que estaba en marcha una revisión de los trabajos e investigaciones que condujeron a la concesión del Premio Nobel en 2016 sobre la significación de la autofagia como sistema clave de la salud en la eliminación y reciclaje de componentes celulares, procesos clave para prevenir la aparición de ciertas patologías crónicas y permitir envejecer de forma más saludable.
Es importante remarcar que este artículo dedicado a la autofagia no pretende hacer una revisión exhaustiva de estos aspectos, aunque sí recordar que las investigaciones que condujeron al Premio Nobel en 2016 han cambiado el concepto de cómo la célula es capaz de reciclar algunos de sus materiales metabólicos y componentes celulares en respuesta a la inanición y el estrés. Sin embargo, en este trabajo especial se revisan primero los aspectos burocráticos que concernieron al Nobel en 2016(6), para posteriormente señalar en qué consiste la autofagia y cuál es su importancia fisiológica.
El Premio Nobel de Fisiología y Medicina de 2016. Burocracia y boato. Autofagia
Cada 10 de diciembre, fecha de la muerte de Alfred Nobel, tal como lleva haciéndose desde 1901, excepto en los años marcados por las dos grandes guerras Mundiales, tiene lugar primero en Oslo la ceremonia de entrega del Premio Nobel de la Paz y unas horas después en Estocolmo para el resto de los Premios Nobel(6,7). Estos premios, instituidos por el químico e inventor Alfred Nobel en su testamento, y organizados y administrados desde hace más de un siglo por la Fundación que lleva su nombre, es uno de los galardones más prestigiosos del mundo y se concede tal como figura en la página oficial del Premio «For the greatest benefits to humankind» (traducción libre al castellano «Para mayor beneficio de la humanidad»(1-3).
El galardón del Premio Nobel demanda un exhaustivo proceso de nominación y selección donde intervienen numerosas personas e instituciones de reconocido prestigio en los diferentes campos que abarcan este premio y en particular en el de Fisiología o Medicina, del que nos ocupamos en esta breve revisión. Grosso modo, el procedimiento comienza en septiembre del año previo a la concesión, cuando el Comité del Nobel de Fisiología o Medicina envía de forma confidencial invitaciones a renombrados profesionales miembros del Instituto Karolinska, profesores universitarios de Fisiología o Medicina en los países aledaños, y miembros de la Real Academia Sueca de las Ciencias para que propongan candidatos que puedan optar al galardón en esta modalidad. Las propuestas de los candidatos se envían posteriormente al Comité justo antes de terminar el mes de enero del año de concesión del Nobel, y durante el mes de febrero, dicho organismo examina los nombres propuestos. Entre los meses de marzo y mayo, dicho Comité consulta sobre la adecuación de los candidatos preliminares y emite un informe que recoge todos los antecedentes e información de la proposición y lo remite a la Asamblea del Nobel, la cual debate sobre estos aspectos en dos reuniones, sometiéndose a principio de octubre a votación de sus miembros la decisión inapelable sobre la designación de los elegidos ganadores(5).
El tres de octubre de 2016 el Secretario del Comité del Nobel de Fisiología y Medicina, el profesor Thomas Perlmann, hacía público a los medios el siguiente comunicado (sic) «The Nobel Assembly at Karolinska Institutet has today decided to award the Nobel Prize in Physiology or Medicine 2016 to Yoshinori Ohsumi for his discoveries of mechanisms for autophagy» (traducción libre «El comité de la Fundación Nobel en el Instituto Karolinska ha decidido hoy conceder el premio Nobel de Fisiología o Medicina a Yoshinori Ohsumi por su descubrimiento de los mecanismos de autofagia»)(6) (Figura 1).

Figura 1. Anuncio del Premio Nobel de Fisiología o Medicina 2016 concedido a Yoshinori Ohsumi por el profesor Thomas Perlmann, secretario del Comité Nobel de Fisiología o Medicina, el 3 de octubre de 2016.
Posteriormente en la ceremonia de entrega de los premios(6,7), acontecida el diez de diciembre de 2016 en Estocolmo, fecha del aniversario del fallecimiento de su fundador (Figura 2), Yoshinori Ohsumi recibió la medalla Nobel, un diploma y un documento acreditando el logro de manos del rey Carlos XVI Gustavo de Suecia. En dicha ceremonia el profesor Nils-Goran Larsson, miembro de la Asamblea Nobel en el Instituto Karolinska y Miembro del Comité Nobel de Fisiología o Medicina pronunció el discurso de presentación del premiado con el Premio Nobel de Fisiología o Medicina 2016, terminando con las siguientes palabras: “Yoshimori Ohsumi, su innovadora investigación ha resuelto un enigma en biología y ha revelado mecanismos para que nuestras células puedan reciclar sus componentes y combatir la enfermedad. Con una serie de brillantes experimentos ha abierto un nuevo campo en biología. En nombre de la Asamblea Nobel del Instituto Karolinska, tengo el gran privilegio de transmitirle nuestras más calurosas felicitaciones. Ahora le ruego que dé un paso adelante para recibir su Premio Nobel de manos de Su Majestad el Rey”.

Imagen obtenida de https://news.culturacolectiva.com/noticias/comienza-la-entrega-de-los-premios-nobel-ya-entregaron-el-de-medicina/
Figura 2. El laureado Yoshinori Ohsumi después de recibir la medalla y el diploma Medalla del Premio Nobel de Medicina.
Como detalle, hay que comentar que el premio que recibió Yoshinori Ohsumi estuvo dotado con ocho millones de coronas suecas (unos 880.000 euros). También, que en el anverso de la medalla que recibió se muestra la efigie de Alfred Nobel con las fechas de su nacimiento NAT MDCCCXXXIII y muerte OB MDCCCXCVI. En el reverso se encuentra representado al “Genio de la Medicina” sosteniendo un libro abierto en su regazo, y recogiendo agua que sale de una roca para saciar la sed de una mujer enferma (Figura 2). La inscripción “Inventas vitam juvat excoluisse per artes” ha sido tomada de la obra La Eneida de Virgilio del siglo I AC. El nombre del Laureado se muestra debajo de la imagen, y además aparece un texto “REG. UNIVERSITAS. MED. CHIR. CAROL”. Diseñada por Erik Lindberg(8).
Detalles Biográficos de Yoshinori Ohsumi
Según recoge la publicación de Cascales Angosto y col.(8) Yoshinori Oshumi nació en Fukuoka, Japón en 1945. Toda su infancia estuvo marcada por las penalidades y privaciones que sufrió Japón después de la Segunda Guerra Mundial. En 1963 se matriculó en la Universidad de Tokio, centrando sus estudios en biología molecular. Después de graduarse como doctor, se dedicó al estudio de los mecanismos de iniciación del ribosoma en E. coli y la acción de la colicina E3. En 1974 se trasladó a la Universidad Rockefeller de Nueva York donde estudió bajo la dirección del Dr. Edelman la fertilización in vitro en el ratón. Posteriormente, inició en levaduras el estudio de la replicación del ADN. En 1977, vuelve a Japón y es contratado como profesor auxiliar en la Facultad de Ciencias de Tokio, donde centra su investigación en el estudio de la membrana vacuolar en levaduras. En 1988, como profesor asociado de la Facultad de las Artes y las Ciencias de la Universidad de Tokio, crea su propio laboratorio e investiga en levaduras sobre los mecanismos fisiológicos implicados en la autofagia con ayuda de la microscopía óptica y electrónica. También estudian los procesos celulares que acontecen en levaduras mutantes en las que la autofagia no tiene lugar.
El equipo de Oshumi encontró, en una primera instancia, quince genes esenciales que intervienen en la autofagia inducida por ayuno, y comenzó a clonar genes relacionados con la autofagia (en inglés. autophagy-related genes), cuyo acrónimo ATG fue mundialmente aceptado. Posteriormente, descubrió la funcionalidad y relación concatenada de las proteínas codificadas por genes ATG, y de los mecanismos moleculares de las proteínas codificadas por los genes ATG de levaduras. Desde 2009, dirige en el Instituto de Tecnología de Tokio investigaciones sobre la dinámica de las membranas en la formación del “autofagosoma” y la relevancia de este proceso en el marco de la biología estructural, celular y molecular.
El mismo doctor Oshumi refiere que la elección de este tema de investigación fue una gran contribución al conocimiento científico, identificando los genes centrales de la autofagia. Actualmente se publican miles de artículos al año en torno a su línea de investigación. Aunque son muchas las publicaciones del grupo del Dr. Oshumi, resaltaremos algunas donde se resalta la importancia de la autofagia en levaduras(9-12). Particularmente, este grupo de investigación está investigando recientemente en los mecanismos de autofagia relacionados con la eliminación de ácido ribonucleico mensajero (ARNm)(12). Según este galardonado con el Nobel, aunque el papel de la autofagia en la degradación de proteínas y orgánulos está bien caracterizado; sin embargo, la degradación del ARN por autofagia en respuesta al estrés y la de ARNm específicos permanece sin ser prácticamente estudiado. En sus propias palabras (sic) “Proponemos que la autofagia actúa como un sistema de degradación del ARNm en el paso de la traducción, agregando un mecanismo para la regulación transcripcional y la inhibición de la traducción global durante la adaptación celular a las condiciones de estrés. La degradación autofágica de los ARNm asociados a los ribosomas en la fase temprana de la respuesta al estrés puede ser necesaria para facilitar el cambio a la traducción de los genes de respuesta al estrés. La degradación de los ARNm involucrados en la asociación de ribosomas puede ser contraria a la intuición, ya que esto puede sugerir una adaptación ineficiente al estrés cuando la célula es menos capaz de tolerar el desperdicio de recursos. Sin embargo, tal degradación probablemente refleje un intrincado equilibrio entre la expresión de los genes diana y la inhibición de la expresión innecesaria de los ARNm diana. Nuestros resultados demuestran que la autofagia está involucrada en la regulación de la expresión génica a través de la degradación del ARNm, por lo que desempeña un papel fundamental en la homeostasis celular”. Efectivamente en ese último estudio se demuestra la degradación selectiva del ARNm por autofagia inducida por rapamicina en levaduras(12). El perfil de ARNm encontrado en las vacuolas revela que subconjuntos de ARNm, como los que codifican la biosíntesis de los aminoácidos y proteínas ribosómicas, se entregan preferentemente a la vacuola para su degradación por autofagia. También se observa que la degradación del ARNm mediada por autofagia está estrechamente acoplada a la traducción en los ribosomas.
Aunque entra en el campo de la especulación, la degradación del ARNm podría tener una enorme importancia en la eficacia de la inmunización de las nuevas vacunas ARNm frente a SARS-CoV-2, ya que tanto el estado nutricional como las posibles características genéticas de las personas inmunizadas condicionarían la eliminación (temprana o tardía) de los ARNm dirigidos para poner en marcha la síntesis de la proteína S del SARS-CoV-2.
Autofagia. Generalidades y Tipos
Autofagia es el término (procedente de las palabras griegas “auto”' que significa “uno mismo” y “phagy” que significa “comer”) adoptado para describir el conjunto de reacciones moleculares que resultan en la degradación de los componentes intracelulares en los lisosomas. Este proceso fue descrito por primera vez en 1963 por Christian de Duve mientras observaba cómo unas pequeñas vesículas denominadas lisosomas ayudaban a la limpieza celular, lo que le llevó a recibir el Premio Nobel de Fisiología o Medicina en 1974, junto con Albert Calude y George Palade. En la década de los 90, Yoshinori Oshumi describió un conjunto de genes relacionados con la autofagia, conocidos como ATG que, junto con sus intentos de identificar la base genética de dicho proceso, le condujeron a recibir el Premio Nobel de Fisiología o Medicina en 2016(13).
El reciclaje es una de las bases de la supervivencia celular, ya que permite reducir los desechos celulares, preservar la energía celular y adaptarse a los diferentes cambios al regular la abundancia de los componentes intracelulares. A diferencia de lo que se creía antiguamente, la autofagia no solo se basa en un proceso de reciclaje de proteínas, sino que contribuye a mantener un balance energético positivo a través de la degradación y utilización de organelas, glucógeno o lípidos. A su vez, también se ha observado la participación de la autofagia en diversos procesos como la diferenciación celular, la remodelación de tejidos, el control del crecimiento y la defensa celular(14).
Descubrimiento de los lisosomas. La antesala del Premio Nobel de 2016
En la década de 1950, el bioquímico Christian de Duve, investigando la acción de la insulina, trató de localizar en el interior de la célula diferentes actividades enzimáticas. Tras diferentes estudios se concluyó que las enzimas proteolíticas, se encontraban secuestradas en el interior de una estructura de membrana a la que de Duve denominó lisosoma(15,16). Christian de Duve y Albert Claude, junto con George Palade, fueron galardonados en 1974 con el Premio Nobel en Fisiología o Medicina por sus descubrimientos relativos a la estructura y organización funcional de la célula(8).
Poco después del descubrimiento de los lisosomas, otros investigadores observaron porciones de citoplasma secuestradas en el interior de unas estructuras membranosas en células renales de ratones con desarrollo normal. También en rata con hidronefrosis se encontraron en células del túbulo proximal estructuras vacuolares que contenían restos de citoplasma y mitocondrias. Estas vacuolas aumentaban notablemente en número a medida que progresaba la degeneración y acumulaban gránulos que contenían fosfatasa ácida. También se observó en hígado de ratón, estructuras de membrana que tenían en su interior porciones de citoplasma degenerado y su cantidad se elevó considerablemente después de perfundir el hígado con glucagón o mediante la exposición a agentes tóxicos.
Reconociendo que las estructuras encontradas tenían la capacidad de digerir el contenido intracelular, Christian de Duve acuñó en 1963 el término autofagia, y discutió ampliamente este concepto en un artículo de revisión publicado unos años más tarde(16,17).
La prueba más convincente de la existencia de la autofagia en células de mamíferos la proporcionaron los resultados obtenidos por microscopía electrónica. Se supo entonces que la autofagia se produce en casos de nivel metabólico basal bajo y se eleva durante la diferenciación y remodelación en una gran variedad de tejidos, tales como cerebro, intestino, riñón, pulmón, hígado, próstata, piel y glándula tiroides. Además, se observó que la autofagia se producía en una amplia gama de células eucariotas, lo que señalaba que era una función conservada a lo largo de la evolución. Una serie de experimentos que combinaban el fraccionamiento subcelular, la autorradiografía y la microscopía electrónica, proporcionaron evidencias de que las fases iniciales de la autofagia incluían la formación de una estructura de doble membrana, el fagóforo, que se extendía alrededor de una porción del citoplasma y se cerraba formando una vesícula, el autofagosoma (Figura 3), que carecía de enzimas hidrolíticos.
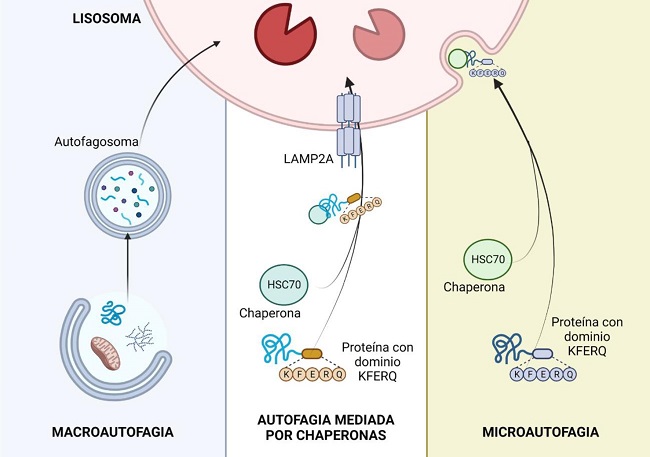
Figura 3. Rutas autofágicas en mamíferos (Modificado de Susmita Kaushik y Ana María Cuervo. The coming of age of chaperone-mediated autophagy)(20).
Al encontrar de Duve que el glucagón inducía en hígado la autofagia, se puso de manifiesto el papel fisiológico de este proceso(18). La observación de que la autofagia se inhibía en presencia de nutrientes y se inducía por inanición, reforzó su estrecha regulación por los nutrientes. Pfeifer y col.(19) encontraron que la inanición durante 48 horas producía la degradación del 30-40% de las proteínas hepáticas. Estos y otros experimentos mostraron que la degradación de las proteínas, vía autofagia, estaba estrictamente controlada por la concentración de determinados aminoácidos en el medio, que la suprimían. Se observó también que la autofagia estaba regulada por hormonas, fenómeno de gran importancia que daba cuenta de su mecanismo de acción. El efecto estimulador del glucagón se oponía al efecto inhibidor de la insulina(8). Con el reciente desarrollo de la espectroscopía de masas, es posible hoy día analizar el comportamiento dinámico de diversos metabolitos. Sin embargo, quedan por demostrar muchos detalles de la regulación y la estrecha relación entre la autofagia y el metabolismo celular.
Casi 30 años después de los descubrimientos de los lisosomas y de acuñar el término autofagia por Christian de Duve, muchos aspectos sobre la autofagia eran mal conocidos, en particular por el desconocimiento de muchos mecanismos moleculares que participaban en la maquinaria autofágica. Así, no se habían identificado aún las proteínas necesarias para la autofagia, ni las que se localizaban en el autofagosoma, con lo que no se disponía de un marcador específico del autofagosoma o del autofagolisosoma para los estudios morfológicos y bioquímicos. Se necesitaba por tanto un buen modelo para estudiar el mecanismo molecular de la autofagia. Este modelo resultó ser la levadura Saccharomyces cerevisiae y que sirvió enormemente para el conocimiento de los mecanismos moleculares implicados en la autofagia desarrollados por Ohsumi(9). Además, muchas incógnitas sobre el mecanismo de autofagia estaban aún sin respuesta (p.ej. ¿Era la autofagia un mecanismo universal de limpieza intracelular? ¿Existen diferentes tipos de autofagia? ¿Tiene la autofagia importancia para la supervivencia del organismo? ¿Juega algún papel en la enfermedad humana?).
Tipos de autofagia
De acuerdo con la forma en que los sustratos alcanzan la luz lisosómica, se han descrito tres formas principales de autofagia en células de mamíferos: macroautofagia, microautofagia y autofagia mediada por chaperonas(20,21) (Figura 3).
Macroautofagia
La macroautofagia es la vía de reciclaje lisosomal mejor caracterizada, en la que los elementos citosólicos destinados a su degradación (carga), son secuestrados por una vesícula de doble membrana que administra el material citosólico a los lisosomas(22,23). Dicha membrana aislante de origen no lisosomal o fagóforo naciente, se forma a través del ensamblaje de proteínas y lípidos procedentes de diferentes orgánulos como el retículo endoplásmico, aparato de Golgi o membrana plasmática. El cierre de la vesícula con la carga en su interior da lugar a lo que se conoce como vacuola autofágica o autofagosoma, que se desplaza a través de los microtúbulos para fusionarse con los lisosomas, orgánulos que proporcionan las enzimas necesarias para la degradación de los componentes secuestrados. Tras la hidrólisis, los aminoácidos, lípidos y restos de hidratos de carbono, alcanzan el citosol a través de transportadores y permeasas para su reciclaje.
La identificación de los genes ATG permitió conocer en más detalle el proceso autofágico. Hasta la fecha los estudios genéticos realizados en levaduras han permitido caracterizar más de 30 genes ATG, muchos de los cuales presentan ortólogos en eucariotas superiores. Una vez que la señal de activación de la autofagia se transduce, los genes ATG controlan cada uno de los pasos de la vía de autofagia, desde la formación del autofagosoma hasta la degradación y reciclaje de la carga(22, 23).
Autofagia mediada por chaperonas
La autofagia mediada por chaperonas (CMA, de sus siglas en inglés chaperone-mediated autophagy) fue descubierta por J. Fred Dicen en 1985 y es la vía autofágica por la que se degradan un subconjunto específico de proteínas que atraviesan la membrana lisosomal a través de la proteína de membrana asociada al lisosoma tipo 2A (LAMP-2A)(22). En este proceso, las proteínas citosólicas destinadas a su degradación son reconocidas por la proteína relacionada con el choque térmico citosólico de 70 kDa (también conocida como chaperona hsc70) a través de un dominio pentapeptídico (KFERQ) de la secuencia de aminoácidos de la proteína. Una vez que el complejo chaperona-sustrato alcanza la membrana lisosomal mediante la unión a LAMP-2A monoméricas, se induce su multimerización en un complejo de translocación que transporta la proteína del sustrato al interior del lisosoma para su degradación. Posteriormente, desplegado e internalizado el LAMP-2A se degrada rápidamente en la luz lisosomal, regulando los niveles de dicho receptor y, por tanto, limitando la actividad de CMA. La activación de este tipo de autofagia, además de actuar en respuesta a los cambios nutricionales, también funciona como mecanismo de defensa contra los ataques celulares en un esfuerzo por eliminar las proteínas dañadas y garantizar una homeostasis proteica adecuada(20-23).
Microautofagia
En este tipo de autofagia las cargas son secuestradas por invaginación directa o protusión del lisosoma, originándose pequeñas vesículas de pared simple a partir de la membrana lisosomal(24). Recientemente, se ha encontrado una variación de este proceso denominada microautofagia endosomal, la cual tiene lugar en endosomas tardíos e implica una degradación selectiva mediante el reconocimiento del dominio KFERQ de proteínas por hsc70, al igual que ocurre en CMA. Sin embargo, la internalización del sustrato se lleva a cabo a través del complejo de clasificación endosomal requerido para el transporte I y III (ESCRTI y III) en lugar de LAMP-2A(25).
Aspectos moleculares centrales de la autofagia
Red mTOR, proteínas y genes AGT
Existen diferentes aspectos por los que una célula aumenta los niveles de autofagia. Entre ellos se encuentran el mal plegamiento de proteínas, la presencia de organelas dañadas que funcionan de modo anómalo o que aumentan su número de modo inadecuado, y una carencia de nutrientes o hipoxia (Figura 4).

Modificado de Costas MA y Rubio MF(28)
Figura 4. La red de señalización mTOR consta de dos vías principales, cada una mediada por un mTORC. La vía mTORC1 sensible a rapamicina controla varios caminos que colectivamente determinan la masa o tamaño de la célula. mTORC1 y mTORC2 responden a factores de crecimiento (insulina / IGF), al estado energético de las células, nutrientes (aminoácidos) y a estrés. mTORC1, compuesto de diferentes proteínas es multimérico, aunque se esquematiza como monómero. Las flechas representan la activación, mientras que las barras representan la inhibición. PI3K (fosfatidilinositol 3 quinasa), Akt/PKB (proteína quinasa B), PDK (proteína quinasa 1-dependiente de fosfatidilinositol), TSC (tuberous sclerosis complex) 1 y 2, LKB1 (serina treonina quinasa codificada por el gen LKB1), AMPK (quinasa dependiente de adenosina monofoasfato), Rheb (RAS homologue enriched in brain) es una GTPasa, mLST8 (proteína letal 8 de mamíferos con SEC13), RAPTOR (regulatory-associated protein of mTOR).
Como se ha comentado la vía metabólica del mTOR (de sus siglas en inglés mammalian target of rapamycin) juega un papel importante en el proceso autofágico. La red de señalización mTOR consta de dos “vías” principales, cada uno mediado por un mTOR.
La vía mTOR1 es inhibida por la rapamicina que controla varios caminos que, determinan la masa o tamaño de la célula, mientras que la vía mTOR2 no sensible a rapamicina controla la síntesis de actina del citoesqueleto y, por tanto, la forma de la célula. La vía mTOR1 es muy importantes en el control de la autofagia y tiene un rol fundamental censando carencia de nutrientes, hipoxia y balance metábólico(26,27) (Figura 4).
El proceso de macroautofagia puede dividirse en fases: 1) nucleación, 2) elongación, 3) formación del autofagosoma maduro, 4) fusión, 5) degradación, y 6) reciclaje (Fig. 5). La fase de nucleación está regulada por el complejo mTORC1, un complejo formado por 5 proteínas: mTOR, Raptor (regulatoryassociated protein of mTOR), GbetaL o mLST8 (proteína letal 8 de mamíferos con SEC13), PRAS 40 (substrato de 40 kDa de Akt rico en prolina) y DEPTOR, cuya regulación depende de la relación AMP/ATP la cual, a su vez, está relacionada con el estado nutricional y metabólico (Fig. 4).

(Creado con BioRender.com)
Figura 5. Génesis y desarrollo del autofagosoma y participación de las diferentes proteínas ATG en sus 6 etapas (nucleación, elongación, formación del autofagosoma maduro, fusión, degradación, y reciclaje. Se inicia por activación de complejo quinasa-quinasa ULK1/2 (proteínas quinasa tipo uridina 1 y 2, respectivamente), que fosforila a la proteína Beclin1 (también conocida como ATG6) que forma parte del complejo iniciador de la nucleación del fagóforo y posteriormente la conjugación del mismo con el complejo ATG5/ATG12/ATG16 (proteínas iniciadoras de autofagia 5, 12 y 16). Continúa con el procesamiento de LC3 (microtubuleassociated protein light chain 3) y la inserción en la membrana del fagóforo. Así se produce la formación del autofagosoma que luego se fusiona con el lisosoma (autofagolisosoma) donde ocurre la degradación del substrato cuyos productos son liberados al citoplasma. Modificado de Costas MA y Rubio MF(28).
Este complejo y las proteínas “río abajo” se regulan por fosforilación/desfosforilación y activan las proteínas encargadas de la iniciación del proceso, vía activación de UKL1/UKL2 (proteínas quinasa tipo uridina 1 y 2, respectivamente) (Fig. 5). Gran parte del proceso es mediado por las proteínas de la familia ATG (proteínas iniciadoras de autofagia), las cuales se unen a las proteínas u organelas dañadas, marcándolas para la formación de fagóforos (vesículas de doble membrana que contienen el material a reciclar). Algunas proteínas Atg se unen a los fagóforos, promoviendo su maduración y fusión con el lisosoma para formar el autofagosoma(29). Esta fusión permite que las enzimas lisosomales se ocupen de la degradación enzimática de los sustratos, cuyos productos (aminoácidos, lípidos) son luego exportados al citoplasma para su reutilización (Fig. 5).
Como vemos en la figura 5, la presencia de las proteínas ATG es esencial para un buen funcionamiento del proceso de reciclado celular, el cual se ha observado juega papeles de indiscutible importancia en la salud y estando las alteraciones del proceso autofágico implicadas en la fisiopatología de las enfermedades cardiovasculares, enfermedades infecciosas, enfermedad de Crohn y trastornos neurodegenerativos(30,31).
Tabla 1. Proteínas ATG y características funcionales.
| Proteínas ATG |
Homólogos en mamíferos | Características y Propiedades Funcionales |
|---|---|---|
| Atg1 | ULK1/2 | Iniciación de la autofagia |
| Atg2 | Atg2A/B | Proteína que interactúa con ATG18 |
| Atg3 | Atg3 | Similar a la enzima E2 para la conjugación en Atg8; regulación de la homeostasis mitocondrial |
| Atg4 | Atg4A-D | Proteasa rica en cisteína; activación y delipidación de ATG8 |
| Atg5 | Atg5 | Formación/elongación de autofagosoma; inducción de apoptosis |
| Atg6 | Beclin 1 | Esencial para la formación del autofagosoma; diana de proteínas de virus para interferir con la función y eficacia de la autofagia |
| Atg7 | Atg7 | Similar al enzima E1 en la conjugación de Atg8 y Atg12 |
| Atg8 | LC3A/B/C | Modificador similar a ubiquitina; conjugación a la membrana del autofagosoma; cierre y determinación del tamaño del autofagosoma; funciones independientes de la autofagia |
| Atg9 | Atg9A/B | Proteína transmembrana; posible "transbordador de membrana” o lanzadera |
| Atg10 | Atg10 | Similar al enzima E2 en la conjugación Atg12 |
| Atg12 | Atg12 | Modificador similar a ubiquitina |
| Atg13 | Atg13 | Iniciación de la autofagia |
| Atg14 | Atg14 L | “Objetivo” del complejo Vps34 |
| Atg16 | Atg16L1/L2 | Complejo Atg12- Atg5/ Atg16L1, esencial para formación del autofagosoma |
| Atg17 | FIP-200 | Iniciación de la autofagia |
| Atg18 | WIPI1-4 | Unión a PIP3; posibles funciones en la formación del omegasoma y /o estadios tardíos de la autofagia |
Desde el descubrimiento de los genes relacionados con la autofagia (ATG) mediante estudios genéticos de la levadura hasta la fecha, se han identificado más de 35 genes ATG en la levadura de los cuales 16 están presentes en los seres humanos (ATG2A, ATG2B, ATG3, ATG4A, ATG4B, ATG4C, ATG4D, ATG5, ATG6 (BECN1), ATG7, ATG9A, ATG9B, ATG10, ATG12, ATG16L1 y ATG16L2)(34). El funcionamiento correcto de la autofagia depende de la presencia de polimorfismos en los genes ATG, pudiendo hablarse de genes candidatos a ser estudiados prioritariamente en errores en la autofagia para los genes ATG 5,12 y ATG16L-1.
En estudios de asociación genética se han descrito que defectos en la autofagia confieren susceptibilidad a varias enfermedades autoinmunes e inflamatorias, en particular a la enfermedad inflamatoria intestinal(35). La presencia de estas proteínas depende de la expresión génica de los genes ATG. Por tanto, la lucha contra muchas enfermedades degenerativas, incluidas diferentes tipos de tumoraciones o cáncer comienza con el conocimiento del transcriptoma autofágico (conjunto de RNAm que se originan a partir del DNA y que van a ser traducidos en los ribosomas), dando lugar a la producción de un conjunto de proteínas (Proteoma). Indiscutiblemente, la expresión génica depende de estímulos individuales, pero hay situaciones en que la expresión génica estaría modulada por efectos epigenéticos no demasiado bien conocidas.
No obstante, nos gustaría señalar que, en la mayoría de las células, la autofagia se produce a niveles basales bajos, pero en condiciones especiales (p.ej. estrés) el proceso aumenta como una respuesta citoprotectora esencial para mantener la supervivencia celular. También se ha sugerido que la supervivencia de células cancerígenas puede estar garantizada por la sobreactivación de la autofagia in vivo y contribuye a la resistencia a quimioterapias y estrés, promoviendo la metástasis y los periodo de latencia(36).
Autofagia, envejecimiento y enfermedades crónicas
Los descubrimientos realizados en el campo de la autofagia han sentado las bases para conocer su implicación fisiológica en una variedad de estados fisiológicos y patológicos. Este proceso se encuentra muy unido a procesos como la embriogénesis, diferenciación celular, adaptación al ayuno y otros tipos de estrés, así como a condiciones patológicas(37). Dado que se han descrito alteraciones de la autofagia en la enfermedad de Alzheimer, cáncer, obesidad, Diabetes Mellitus tipo 2 (DMT2) o enfermedad del hígado graso no alcohólico, parece muy probable que la base de estas patologías resida en fallo de las diferentes rutas autofágicas(14).
Enfermedades neurodegenerativas
La enfermedad de Alzheimer se ha relacionado con fallos en los mecanismos autofágicos(38). Las proteínas mal plegadas tienden a formar agregados insolubles que pueden resultar tóxicos para las células. En modelos de enfermedades degenerativas (p.ej. moscas y ratón) la activación de la autofagia por inhibición de la quinasa TOR reduce la toxicidad de los agregados proteicos. Además, la pérdida de la autofagia en cerebro de ratón, por la disrupción especifica de las proteínas Atg5 y Atg7 causa neurodegeneración. En una revisión reciente se relacionan algunos procesos de la autofagia y su implicación en la enfermedad de Alzheimer, analizando el papel de la vía del mTOR, de la neuroinflamación, de los endocannabinoides, y de algunos genes como el ATG7, BCL2, BECN1, CDK5, CLU, CTSD, FOXO1, GFAP, ITPR1, MAPT, PSEN1, SNCA, UBQLN1, and UCHL1. De forma similar se ha relacionado la autofagia con el acúmulo de proteínas como la s-sinucleína y de neuronas dopaminérgicas degeneradas en la enfermedad del Parkinson(39).
Enfermedades metabólicas
La gran mayoría de los estudios señalan que en la fisiopatología de la DMT2 participan alteraciones en el hígado, páncreas y tejido adiposo. Estas alteraciones están relacionadas con modificaciones en el proceso autofágico. Así, ratones knockout para Atg7, un gen esencial para la actividad de macroautofagia, mostraron defectos estructurales y funcionales en las células β pancreáticas, que condujo a intolerancia a la glucosa(40,41). Esta misma deleción en el hígado cursó con un mayor tamaño de hígado y acumulación de triglicéridos como consecuencia de una desregulación de la lipofagia; mientras que en tejido adiposo provocó una menor formación de depósitos grasos por una diferenciación defectuosa de los adipocitos(42). Resultados similares se han encontrado para ratones knockout de LAMP-2 hepática, proteína de membrana asociada a los lisosomas que regula CMA. Estos animales presentaron mayor acumulación de lípidos hepáticos debido a un gran aumento de las enzimas hipogénicas(43).
En el inicio de la DMT2, las células β-pancreáticas se adaptan a la situación de resistencia a la insulina, incrementando la producción y secreción de insulina. Esta constante demanda se ha asociado a un mayor acúmulo de proteínas mal plegadas, especialmente proinsulina, que provocan un estrés de retículo endoplasmático(44). La continua captación de glucosa por parte del páncreas y su posterior metabolismo, inducen una elevada cantidad de especies reactivas de oxígeno que no pueden ser contrarrestada por la maquinaria antioxidante. Asimismo, la llegada de ácidos grasos libres y citoquinas proinflamatorias de los tejidos periféricos también contribuye y agravan el estrés celular, provocando la disfunción y el fallo de la célula β-pancreática(45). La autofagia puede prevenir el estrés celular al eliminar las proteínas mal plegadas y orgánulos no funcionales como mitocondrias dañadas. Sin embargo, en la situación diabética la actividad autofágica puede estar reducida o alterada por estimulación por glucosa y ácidos grasos libres de mTORC1, respectivamente (Figura 5) o por inhibición de la acidificación lisosómica (ácidos grasos libres y acumulación de amilina), lo que conduce a la destrucción de las células β y contribuye al círculo vicioso de la DMT2(44). A pesar de todos los indicios que asocian la DMT2 con una alteración de la autofagia, todavía no hay evidencia clara y se requieren estudios adicionales para conocer si la desregulación de la actividad autofágica es causa o consecuencia de dicha patología.
Cáncer
Se ha encontrado asociación de la autofagia con el cáncer en dos momentos cruciales: uno en las primeras etapas del desarrollo, donde el control de la autofagia, particularmente sobre el mantenimiento del genoma, inhibiendo la tumorigénesis y confiriéndole funciones antioncogénicas. Así, la autofagia podría coordinar el mantenimiento o entrada de las células en fase G0 y, en consecuencia, prevenir la hiperproliferación espontánea. Y otro, centrado en el tratamiento: en las últimas etapas del cáncer, en el que la autofagia puede ayudar a que las células tumorales sometidas al estrés metabólico resistan la muerte desencadenada por quimioterápicos (46).
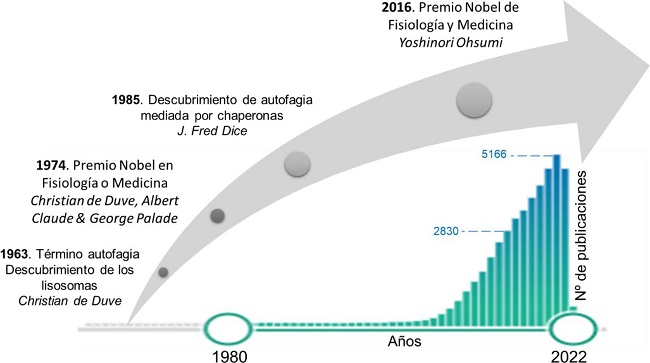
La figura 6 resume los aspectos claves acontecidos desde el descubrimiento de los lisosomas por de Duve en 1983 hasta 2016, en el que se concedió el galardón del premio Nobel a Yoshinori Ohsumi. Desde 2016, han tenido lugar grandes avances, particularmente en la aplicación de los estudios a la protección frente al envejecimiento y las enfermedades degenerativas, sin olvidar el estudio de fármacos para la lucha de enfermedades como el cáncer.
No obstante, en publicaciones posteriores en JONNPR nos gustaría resaltar la personalidad y el trabajo realizado en el campo de la autofagia por algunos investigadores españoles como la Dra. Ana María Cuervo.
Fármacos y autofagia
El conocimiento del papel de las proteínas Atg en la autofagia ha potenciado enormemente la investigación farmacológica de la autofagia en relación principalmente con el cáncer. Como hemos señalado en la mayoría de las células la autofagia se produce a niveles basales bajos, pero en condiciones especiales (p.ej. estrés) el proceso aumenta como respuesta citoprotectora esencial para mantener la supervivencia celular. También se ha sugerido que la sobreactivación de la autofagia promueve la supervivencia de células cancerígenas en el microambiente tumoral in vivo y contribuye a la resistencia a quimioterapias y estrés, promoviendo la metástasis y la latencia(36). Por ello el uso de fármacos para activar la autofagia es un aspecto bastante controvertido(47). Así, la cloroquina, se ha utilizado dado que modifica la permeabilidad de la membrana lisosomal, liberando enzimas proteolíticas que inducen daños celulares, que finalmente conducirán a la apoptosis(48).
Por su parte el panobinostat es un potente inhibidor de pan-deacetilasas, ya que puede bloquear múltiples vías relacionadas con el desarrollo tumoral y revertir cambios epigenéticos implicados en la progresión tal como señalan Sharma y col.(49), Su mecanismo conduce a la apoptosis de células malignas a través de diferentes mecanismos.
No obstante, comentaremos que diversos fármacos pueden intervenir en la regulación de la autofagia, la cual puede ser inhibida por 3 metil-adenina, por inhibidores del complejo fosfatidil inositol 3quinasa III (PI3K de clase 3) y por spautin-1, que promueve la degradación de los complejos PI3K de clase III(28,36). La verteporfina pueden interferir, una vez iniciado la formación del fagóforo, con los motivos de la región que interactúan con LC3 (Figura 5) y bloquear el reclutamiento selectivo de cargas (p.ej. mitocondrias)(28,50). La desestabilización de los microtúbulos por los alcaloides de la vinca puede bloquear la maduración de los autofagosomas, mientras que la estabilización por el taxol puede aumentar la fusión entre las vacuolas autofágicas y los lisosomas(28,51). La monensina, cloroquina y NH4Cl, son compuestos que aumentan el pH lisosómico e interfieren con la función lisosómica y bloquean la autofagia en una etapa tardía(28,36,52). La etapa final de la autofagia también puede ser bloqueada por inhibidores de enzimas lisosómicas, tales como E64d, pepstatina A, y bafilomicina A1(28,36). En consonancia con el papel controvertido de los fármacos en la tumorigénesis, se ha señalado que la expresión elevada del coactivador de receptores nucleares el RAC3, generalmente sobreexpresada en varios tipos de cáncer(28,53-57), preserva a la célula tumoral de la muerte por autofagia en la fase de iniciación. Sin embargo, cabe señalar que la hipoxia inhibe la expresión de RAC3, originado una situación permisiva para el proceso de autofagia que preserva la supervivencia de las células tumorales ante la carencia de oxígeno y nutrientes hasta que tenga lugar la angiogénesis(28,58).
Conclusiones
Más de sesenta años han pasado desde el inicio de los estudios de la autofagia. Durante muchos años no se ha considerado que la autofagia era la principal vía de degradación de las proteínas y de los orgánulos subcelulares. En las últimas décadas se han puesto en evidencia muchos de los mecanismos moleculares de la autofagia y su significación fisiológica, con especial mención de los estudios que llevaron al Nobel en 2016 y los realizados por otros grupos. La identificación, por Ohsumi, de los genes ATG en levadura supuso un enorme avance en el conocimiento de la autofagia. Sin embargo, queda mucho por hacer ya que estamos aún en las etapas iniciales del conocimiento de este proceso. Sabemos que la degradación es una función fundamental de la célula para el mantenimiento de la vida, tan esencial como lo es la síntesis. Se necesita profundizar en el conocimiento de la dinámica de las membranas que constituyen la autofagia y avanzar en el desarrollo de sistemas que permitan visualizar la maquinaria dinámica de la autofagia con elevada resolución espacio/temporal. Se necesita más información sobre la composición y localización de moléculas lipídicas en el interior de la membrana del autofagosoma y sobre la estructura bioquímica y los análisis de las proteínas relacionadas con las Atg y los genes que las codifican, habiéndose definido algunos polimorfismos de ATG5/ATG12/ATG16 claves ya que participan en la iniciación de la formación del autofagosoma.
Debido a los trabajos de Yoshinori Ohsumi y Ana María Cuervo, entre otros, la autofagia se reconoce hoy como un proceso fundamental de la fisiología celular con importantes implicaciones en la salud y en la enfermedad.
También es importante reseñar que, bajo condiciones como el estrés, la autofagia aumenta para mantener la supervivencia celular, como una respuesta citoprotectora esencial. De acuerdo con lo expuesto en esta revisión, las alteraciones del proceso autofágico están implicadas en la fisiopatología de cardiomiopatías, enfermedades infecciosas, enfermedad de Crohn y trastornos neurodegenerativos. También se ha sugerido que la sobreactivación de la autofagia puede promover la supervivencia de células cancerígenas en el microambiente tumoral in vivo y contribuye a la resistencia a quimioterapias y estrés, promoviendo la metástasis y la latencia.
Por tanto y para concluir las investigaciones en autofagia, muy potenciadas por la concesión del Nobel a Ohsumi en 2016 han abierto campos de investigación sin precedentes en la lucha contra el envejecimiento y las enfermedades degenerativas que hacen plantearnos, entre otros aspectos, que existe una interacción enorme entre nuestro yo y el ambiente, particularmente en periodos críticos de crecimiento y renovación celular, siendo de enorme y particular importancia el estatus nutricional desde incluso antes del nacimiento(59-61).

